
Abstract
Sudden cardiac death (SCD) is still a major public health issue with an estimated annual incidence ranging from 184,000 to > 400,000 per year. The ACC/AHA/ESC 2006 guidelines define SCD as “death from an unexpected circulatory arrest, usually due to a cardiac arrhythmia occurring within an hour of the onset of symptoms”. A recent study of sudden cardiac death using multiple sources of ascertainment found that coronary artery disease was present in more than 50% of patients older than 35 years who died suddenly and underwent autopsy. Antiarrhythmic drugs have failed to show any mortality benefit even when compared to placebo or implantable cardiovertor defibrillators (ICDs). While patients with systolic heart failure are at higher risk of dying suddenly, most of the patients experiencing sudden cardiac death have left ventricular ejection fraction (LVEF) > 50%. β-blockers, Angiotensin enzymes (ACE) inhibitors as well as aldosterone antagonists prevent ischemia and remodelling in the left ventricle especially in post myocardial infarction (MI) patients and in patients with systolic heart failure. This article will review the data on the effects of traditional heart failure medications, especially β-blockers, Renin Angiotensin system blockers, as well as Statin therapy on sudden cardiac death in post MI patients and in patients with systolic heart failure.
Introduction
Sudden cardiac death (SCD) is still a major public health issue with an estimated annual incidence ranging from 184 000 to > 400 000 per year. 1 –4 The American College of Cardiology/ American Heart Association/ European Society of Cardiology 2006 guidelines define SCD as “death from an unexpected circulatory arrest, usually due to a cardiac arrhythmia occurring within an hour of the onset of symptoms.” 5 The World Health Organization defines SCD as “sudden, unexpected death either within 1 hour of symptom onset (event witnessed) or within 24 hours of having been observed alive and symptom free (un-witnessed).” 6 The SCD estimation is difficult, and there is a wide range reported in the studies. 1 –4 This is due to reliance on death certificates in earlier studies, which can over- or underestimate the incidence of SCD. 7 Furthermore, there is poor agreement between autopsy diagnosis and death certificates. 8 –10 Using prospective and multiple sources of ascertainment including review of medical records, analysis of circumstances of death and autopsy data when available are important methods that can help better estimate the true incidence of SCD. 3,11 But of the 6 original source studies reporting primary data on SCD, only 2 studies were prospective. 3,6,11 A recent study of SCD using multiple sources of ascertainment found that coronary artery disease was present in more than 50% of the patients older than 35 years who had SCD and underwent autopsy. 3 It is also important to note that patients presenting with ventricular fibrillation/ventricular tachycardia (VF/VT) are more likely to survive than patients presenting with asystole or pulseless electrical activity (25% vs 2%, respectively). 3 Antiarrhythmic drugs have failed to show any mortality benefit even when compared to placebo or implantable cardioverter defibrillators (ICDs). 12 Although patients with systolic heart failure are at higher risk of dying suddenly, most of the patients experiencing SCD have left ventricular ejection fraction (LVEF) >50%. Measuring LVEF has its own pitfalls and alone is not sufficient as a risk predictor. 13,14 Relying on the left ventricular (LV) function alone is not enough when making health care policy, especially since majority of sudden cardiac arrests are the first arrhythmic event a patient experiences and could be the first presentation of coronary artery disease. 15 Measuring LV mass and the presence of LV hypertrophy can predict SCD and add to risk stratification even in patients with low LVEF. 16,17 Furthermore, patients with New York Heart Association (NYHA) functional class III and IV systolic heart failure are at high risk of sudden death, 18,19 and exercise tolerance is an important predictor of survival in patients with advanced systolic heart failure independent of LVEF. 20 β-Blockers, angiotensin enzyme (ACE) inhibitors, and aldosterone antagonists prevent ischemia and remodeling in the left ventricle, especially in post-myocardial infarction (MI) patients and in patients with systolic heart failure. This article will review the data on the effects of traditional heart failure medications, especially β-blockers and renin–angiotensin system blockers as well as statin therapy on SCD in post-MI patients and in patients with systolic heart failure.
β-Blockers and SCD Prevention
Potential Mechanisms of β-Blockers on SCD Prevention
Multiple studies have suggested that the major mechanisms responsible for the cardiac arrhythmias associated with SCD are VT and VF. For these arrhythmias to occur, an interaction between substrate (ventricular enlargement and/or hypertrophy and myocardial scar due to ischemic or nonischemic injury) and triggers (electrolyte abnormalities, changes in the sympathetic and parasympathetic activity, neurohumoral factors, and premature ventricular contractions) is necessary to initiate reentry leading to VT and VF (Figure 1). Even in the setting of early MI (<48 hours), VT can occur and is mostly due to reentry caused by the electrical inhomogeneity of the ischemic myocardium. 21 Other mechanisms such as triggered activity and increased automaticity could also be involved. Early VF after MI (<48 hours) can occur without LV dysfunction. Late VF (>48 hours) occurs mainly in the setting of large infarcts, typically in the anterior wall and is mostly thought to be due to reentry. 21 In any reentry circuit, there is an excitable gap. An excitable gap is the region between the head and the tail of the preceding reentrant wave front that is capable of being excited (no longer refractory). The larger the excitable gap, the more likely a stimulus is capable of initiating or terminating the tachycardia. Arrhythmias with smaller excitable gaps are less likely to sustain. 22

Diagram showing the interaction of various substrate and triggers that modulate potential arrhythmogenic mechanisms that lead to ventricular tachycardia and fibrillation, which are the most common arrhythmias causing sudden cardiac death. SCD indicates sudden cardiac death.
Many anatomic or functional substrates such as coronary artery disease, cardiomyopathy, or primary electrophysiological disease can lead to SCD. Progression of these disease states leads to sympathetic activation. At the cellular level, sympathetic and vagal denervation caused by myocardial ischemia leads to an increase in interstitial potassium and intracellular calcium concentrations. 15 Sympathetic activation results in protein kinase A phosphorylation of the ryanodine receptors (RyR2) leading to diastolic release of calcium from sarcoplasmic reticulum, which increases after depolarizations that can be the trigger for ventricular arrhythmias. 23 β1 receptor stimulation can also lead to phosphorylation of the RyR2 receptor via a pathway independent of protein kinase A by activating an exchange protein directly activated by cyclic adenosine monophosphate (cAMP; Epac). This leads to phosphorylation of the RyR2 receptors and calcium leak from the sarcoplasmic reticulum. 24 Blocking β1 receptors prevents this calcium leak. Furthermore, increased intracellular calcium leads to partial membrane depolarization that causes a decrease in the steady state availability of fast sodium channels. 25 The area of damaged or diseased myocardium does not need to be large to create reentry. Often, ischemic myocardium is adjacent to healthy myocardial tissue, which leads to heterogeneity of conduction and creates the substrate that leads to ventricular arrhythmias when a timely trigger (from early or delayed after depolarizations) occurs. 26
As myocardial ischemia progresses, the neurohumoral system exerts further stimulation of the sympathetic system and the renin–angiotensin–aldosterone system (RAAS). This neurohumoral cascade leads to increasing levels of norepinephrine, angiotensin II, aldosterone, endothelin, and vasopressin. Increased norepinephrine levels lead to increased preload and afterload, which in turn increases myocardial oxygen demand. Furthermore, the activation of these systems promotes fibrosis and necrosis, 27 –29 which over time will lead to cardiac remodeling, LV dilatation, fibrosis, and progression into heart failure. 30
Three types of β receptors are known, designated β1, β2, and β3 receptors. β1 receptors are located mainly in the heart and in the kidneys and are downregulated in heart failure due to chronically elevated norepinephrine levels. β2 receptors are located mainly in the lungs, gastrointestinal tract, liver, uterus, vascular smooth muscle, and skeletal muscle. β3 receptors are located in fat cells, and they have activity in the heart. β3 receptor expression in the heart is increased after 4 weeks of exercise, and they are important in regulating endothelial nitric oxide synthase and are necessary for regulating and mediating the cardioprotective effects of exercise. 31 β1 and β2 receptors activate cAMP that acts as a second messenger and leads to increased contractility (inotropy), increased heart rate (which increases myocardial oxygen demand), increased conduction velocity (which may promote reentry), and have a positive lusitropic effect that improves active relaxation. 32 β2 receptors promote the release of renin, which in turn activates angiotensin II and aldosterone, both of which elevate the blood pressure, increase afterload, promote potassium wasting, and activate fibroblasts leading to fibrosis. β-Blockers are considered as class II antiarrhythmic medications. They decrease spontaneous depolarization, prolong the sinus node cycle length, atrioventricular conduction times, and atrioventricular refractory periods. β-Blockers exert their protective effect on the heart via different mechanisms. β-Blockers reduce ischemia by decreasing the heart rate, which is the major determinant of myocardial oxygen demand. 33 At the cellular level, β-blockers decrease electrical excitability by limiting calcium entry via catecholamine-dependent channels. 24,34 They also restore cardiac ryanodine receptor channel function in patients with heart failure, and they upregulate β receptor density. 23 All this helps decrease LV mass and volume, decrease LV end-diastolic pressure, and improve LV and RV function. 23,35 β-Blockers improve LV function over time in patients with systolic heart failure, and this is evident after 1 to 3 months of therapy and continues over time. By 18 months, the LV geometry also improves, and the heart becomes less spherical. 36 However, this does not occur in all patients with systolic heart failure. Several studies suggest that the presence of hibernating myocardium predicts response to β-blocker therapy, and segments that show extensive scarring (>75% of gadolinium enhancement) are less likely to improve. 37,38 In a subanalysis of The Multicenter Automatic Defibrillator Implantation Trial II (MADIT–II) trial, the benefit of implanting an ICD was found to be time dependent, with benefit extending for patients who had a long history of poor LV systolic function and remote MI despite receiving β-blockers. 39 Furthermore, β-blockers have been shown to decrease the frequency of ICD shocks, 40 and they increase the success of antitachycardia pacing. 41 All these data suggest the benefit of β-blockers on survival is not explained only by the improvement in LV function.
Effect of β-Blockers on SCD Prevention in Post-MI Patients
β-Blockers were studied in the post-MI patients since the 1960s when propranolol was found to reduce the mortality after acute MI. 42 Pivotal trials such as the Norwegian Multicenter Study Group (utilizing Timolol at a starting dose of 5 mg/d with target of 20 mg/d), β-blocker Heart Attack Trial (BHAT, utilizing propranolol at a dose of 180-240 mg/d) in the 1980s showed reduction in total mortality and SCD. 43,44 The benefit of β-blockers post-MI was seen even in patients taking ACE inhibitors. The Survival and Ventricular Enlargement (SAVE) and Acute Infarction Ramipril Efficacy (AIRE) trials demonstrated that β-blockers provided additional reduction in cardiovascular mortality independent of the use of ACE inhibitors. 45,46
A meta-analysis evaluated several randomized clinical trials looking at the benefits of β-blockers treatment post-MI. This analysis revealed a significant reduction in mortality with β-blocker therapy (hazard ratio [HR] = 0.77, 95% confidence interval [CI] 0.69-0.85). 47 Despite this, β-blockers are underutilized as was shown by the Cooperative Cardiovascular Project, an observational study that evaluated the care of 200 000 Medicare patients with the diagnosis of MI. Only 34% of the patients were given β-blockers. The mortality reduction in patients who were prescribed β-blockers at the time of discharge from the hospital was 40%. 48
Even patients who have depressed LV systolic function post-MI benefit from β-blockers as shown by a subanalysis of BHAT trial. 49 The Carvedilol Post-Infarct Survival Control in Left Ventricular Dysfunction trial (CAPRICORN) trial specifically addressed this patient population. The CAPRICORN was a prospective, randomized trial recruiting patients with recent acute MI (3-21 days) and LV dysfunction with ejection fraction (EF) ≤40%. A total of 984 patients were placed on placebo and 975 patients were allocated to carvedilol therapy post-MI with an average follow-up of 1.3 years. The initial starting dose was 6.25 mg orally twice daily with target dose of 25 mg orally twice daily. All-cause mortality was lower in the carvedilol group than in the placebo group (HR 0.77, 95% CI 0.60-0.98, P = .03). 50 Patients taking carvedilol had a significant reduction in malignant ventricular arrhythmias (HR 0.37, 95% CI 0.24-0.58; P < .0001). 51 It is important to emphasize that in this trial 98% of the patients were treated with an ACE inhibitor.
Effect of β-Blockers on SCD Prevention in Patients With Systolic Heart Failure
β-Blockers were initially thought to be contraindicated in patients with heart failure due to their negative inotropic effects in the short term. However, later studies showed they consistently improve morbidity and mortality in patients with heart failure; they also lead to a 40% reduction in hospitalization. The first is the US Carvedilol trial that enrolled 1094 patients with congestive heart failure (CHF) and LVEF of ≤35%. Patients were assigned to 4 treatment protocols based on exercise capacity. Within each protocol, patients were assigned to either placebo (n = 398) or carvedilol (n = 696). Although this trial was not designed as a mortality trial, it demonstrated a 65% decrease in the risk of death with carvedilol compared to placebo (P < .001). Sudden death was reduced from 3.8% in the placebo group to 1.7% in the carvedilol group. 52
The Carvedilol Prospective Randomized Cumulative Survival (COPERNICUS) trial examined the effect of carvedilol in 2289 patients with severe CHF, defined as dyspnea at rest and LVEF ≤25%. This trial validated the mortality benefit of carvedilol in patients with severe heart failure with a 50% reduction in all-cause mortality (HR 0.50, 95% CI of 0.10-0.63, P < .01). 53 Unfortunately, this trial did not have data available on the impact of carvedilol on sudden death.
The Cardiac Insufficiency Bisoprolol Study (CIBIS) II was a multicenter, double-blind, randomized, placebo-controlled trial that evaluated the efficacy of bisoprolol in reducing the incidence of all-cause mortality in heart failure. Bisoprolol is a selective β1 receptor blocker, and the target dose was 10 mg daily. All patients enrolled received standard therapy with diuretics and ACE inhibitors (ACE-I). A total of 2647 patients with NYHA class III or IV with LVEF of ≤35% were randomized to either bisoprolol (n = 1327) or placebo (n = 1320). This study was stopped prematurely because bisoprolol showed a significant mortality benefit. Death from any cause in the Bisoprolol group was 11.8% versus 17.3% in the placebo group (HR 0.66, 95% CI 0.54-0.80, P = .0001). Sudden death was also reduced in the bisoprolol group by 42% compared to the placebo group (HR = 0.56, 95% CI 0.39-0.80, P = .0011). 54
The Metoprolol CR/XL Randomized Intervention Trial in CHF (MERIT-HF) is a double-blind randomized controlled study that included 3991 patients with CHF and NYHA class II to IV with an LVEF of ≤40%. These patients were stable on optimal medical therapy. This trial evaluated whether controlled release/extended release formulation of metoprolol taken daily would reduce mortality in this patient population. The starting dose was 12.5 mg once daily with target dose of 200 mg orally once daily. Patients were randomized to metoprolol CR/XL (n = 1990) uptitrated to 200 mg daily over and 8-week period of time or placebo (n = 2001). The trial demonstrated a 34% relative risk reduction in all-cause mortality with controlled release/extended release formulation of metoprolol. The MERIT-HF showed a 41% relative risk reduction in sudden death. 55
The β-blocker Evaluation of Survival (BEST) Trial showed a statistically nonsignificant trend toward reduced mortality with bucindolol in 2708 patients with mild to moderate HF and LVEF of <35% (HR 0.90, 95% CI 0.78-1.01, P = .10). 56 There are significant differences between this trial and CIBIS II, COPERNICOUS, and MERIT-HF trials. When the BEST study population was adjusted to exclude older patients (>80 years of age), patients with systolic blood pressure <100 mm Hg and heart rate <60 bpm (which were exclusion criteria for the other trials), bucindolol treatment was associated with significantly lower risk of death from all causes (HR = 0.77, 95% CI = 0.65-0.92) and sudden death (HR = 0.77, 95% CI 0.59-0.999). 57 This suggests that the differences observed related to the population differences between these trials. However, further research needs to be done to further clarify this issue.
The only β-blockers approved for the treatment of heart failure in the Unites States include carvedilol, metoprolol succinate and bisoprolol. Carvedilol is a third-generation nonselective β-blocker that also blocks α1 receptor and has a 12-fold higher affinity to the β1 receptor compared to metoprolol. Furthermore, it does not upregulate the β receptors in the heart as does metoprolol. It also inhibits smooth muscle mitogenesis and attenuates oxygen-free radical-initiated lipid peroxidation independent of its adrenoceptor-blocking activity. 58 Metoprolol Succinate crosses the blood–brain barrier and rarely can cause nightmares and severe depression. 59 This led some investigators to conclude that carvedilol is superior to metoprolol Succinate in patients with heart failure. However, both agents improve LV function and survival, and both have been shown to decrease hospitalizations as discussed earlier. 53,55,60 It is always best to continue β-blockers during hospitalization for heart failure. In the Organized Program to Initiate Lifesaving Treatment in Hospitalized Patients with Heart Failure (OPTIMIZE–HF) registry, patients who discontinued β-blockers had higher mortality compared to those who continued on β-blockers (HR 2.3, 95% CI 1.2-4.6, P = .013) and had similar risk of death as the patients who were eligible but did not receive β-blockers. 61 There is no difference on the benefit obtained in patients with diabetes (risk ratio [RR] 0.77, 95% CI 0.61-0.96) versus nondiabetic patients (RR 0.65, 95% CI 0.57-0.74) or in black versus white patients. Furthermore, there is an inverse relationship between β-blockers dose and the combined all-cause mortality and all-cause hospitalization. 62 Despite this, only 17.5% and 7.9% of patients were receiving target doses of metoprolol Succinate and carvedilol, respectively, within 90 days of discharge. 63 This could be due to the lack of access to frequent follow-up, to the lack of understanding of the importance of uptitration to maximum dose, to slower titration protocols, or to fear of poor tolerability of higher doses. The guidelines emphasize uptitration to maximum tolerated dose within 90 days of initiating treatment. 64 Table 1 lists the major trials examining the effects of β-blockers in patients with systolic heart failure.
Major Trials Examining the Effects of β-Blockers in Patients With Systolic Heart Failure.
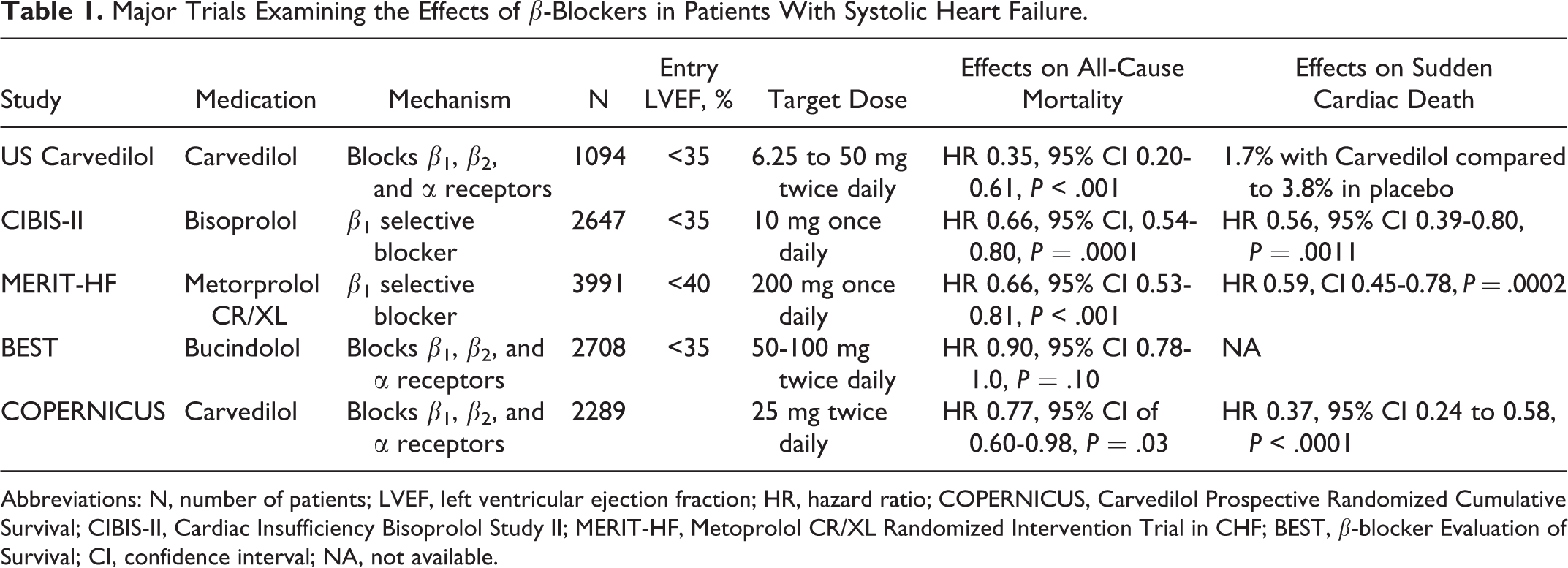
Abbreviations: N, number of patients; LVEF, left ventricular ejection fraction; HR, hazard ratio; COPERNICUS, Carvedilol Prospective Randomized Cumulative Survival;
Effect of β-Blockers on SCD Prevention in Patients Who Survived Cardiac Arrest
In patients who have ICDs, β-blockers have been shown to decrease the frequency of ICD shocks. 40 In an analysis of the Antiarrhythmics Versus Implantable Defibrillators Registry (AVID registry), β-blockers were associated with lower mortality in patients with sustained VT. 65 β-Blockers increase the time to first ICD shock in patients implanted for secondary prevention of sudden death. 66
Furthermore, the higher the dose of β-blockers used, the less patients experience VT and the more likely the therapies are successful. In a study of 282 patients with LV dysfunction (EF < 50%) with standard indications for ICD without cardiac resynchronization therapy, the higher the dose of β-blockers, the more successful antitachycardia pacing was in terminating VT. 41
Effect of β-Blockers on SCD Prevention in Patients With Heart Failure and Normal EF
Patients with heart failure and normal ejection fraction (HFNEF) represent 50% of patients presenting with heart failure. Despite the normal LVEF, these patients have a prognosis similar to patients with systolic heart failure. 67 However, the exact definition has been evolving, and the best and latest definition was proposed by the European Society of Cardiology in 2007. HFNEF was defined by the presence of signs and symptoms of heart failure, normal LVEF of >50%, LV end-diastolic volume index (LVEDVI) <97 mL/m2, signs of diastolic dysfunction on cardiac catheterization or echocardiography. 68 In patients with HFNEF, concentric hypertrophy and high LV mass index to LVEDVI (LVMI/LVEDVI) ratio are present. At the cellular level, there is myocyte hypertrophy, and the cell grows in a transverse direction while keeping the cell length constant. 69 There is also increased deposition of collagen in the interstitial space and loss of connexin 43 expression, which can lead to slow cell-to-cell conduction. 69 It should be noted that most of the patients with HFNEF have history of hypertension, diabetes, and obesity in the absence of hypertrophic cardiomyopathy. 69 Hypertrophied myocytes are more sensitive to ischemia, and ATP-sensitive potassium channels are more likely to open in hypertrophied myocytes compared to normal myocytes. This can lead to prolonged action potential duration, early after depolarizations which can lead to triggered activity and ventricular arrhythmias. 70 Animal models of LV hypertrophy show a lower threshold for VF, especially when myocardial ischemia is present. 71 –74 This could explain the increased mortality and sudden death in patients with coronary artery disease who have LV hypertrophy. 17
Despite the effects of β-blockers on LV hypertrophy and the registry data showing mortality benefit with β-blockers in patients with heart failure and normal EF, 75,76 there is limited clinical trial data available that firmly establish effects on mortality and SCD. The Study of the Effects of Nebivolol Intervention on Outcomes and Rehospitalization in Seniors with Heart Failure (SENIOR) trial randomized 2128 patients aged ≥70 years with a history of heart failure (recent hospitalization with heart failure or LVEF <35%). The primary end point of death or hospitalization was lower with nebivolol compared to placebo in the overall population, but all-cause mortality was not different between the 2 groups. Only a third of the patients enrolled had LVEF >35%, and even in this subset of patients the overall mortality was not different. The study had several limitations since it was not powered enough to detect difference in mortality in patients with LVEF >35%. Furthermore, there was no strict definition for HFNEF in this trial, which makes it difficult to make any recommendations or generalizations in this patient population. 77
Renin–Angiotensin–Aldosterone System and SCD Prevention
Potential Mechanisms of RAAS Inhibitors/Blockers on SCD Prevention
The RAAS is activated during many disease states but especially during myocardial ischemia and heart failure. Renin activates the angiotensin-converting enzyme that converts angiotensin I to angiotensin II. Angiotensin II has 2 receptors, AT1 and AT2. When angiotensin II activates AT1 receptors, it acts as a potent vasoconstrictor and activates fibroblasts leading to interstitial fibrosis and scar formation. Furthermore, AT1 receptor stimulation activates the secretion of aldosterone and norepinephrine. All of these factors increase afterload, which increases myocardial oxygen demand and leads to LV hypertrophy. 78 Angiotensin II increases protein synthesis in cardiac myocytes, and it leads to mitogenesis of cardiac fibroblasts. This is a direct effect mediated by AT1 receptors and is not related to the effects of angiotensin II on blood pressure. 79 Furthermore, angiotensin II induces the angiotensinogen and transforming growth factor β1 genes, which leads to a positive feedback of the cardiac hypertrophic response. 79
Angiotensin II can also be found in the heart tissue and can be synthesized through ACE-independent pathway through the activation of chymase. 80 Chymase has angiotensin-forming capacity that is 20-fold higher than ACE and directly activates metalloproteinases. 80,81 This is not subject to inhibition by ACE inhibitors. The ACE inhibitors and angiotensin receptor blockers (ARBs) block the uptake of angiotensin II in noninfarcted myocardium through AT1 receptors. This leads to attenuation of LV remodeling and also increases local angiotensin II production. By blocking AT1 receptors, the increased angiotensin II activates AT2 receptors. Activation of AT2 receptors leads to vasodilatation and inhibits cell growth and mitogen-activated protein kinases that prevent fibrosis. 82 The antihypertrophic effects of RAAS blockade result not only from diminished AT1 receptor stimulation but also from increased AT2 receptor stimulation. 83
At the cellular level, angiotensin II destabilizes Kv4.3 messenger RNA, a major contributor to outward potassium current. 84 This reduces outward potassium current and leads to prolongation of action potential duration, especially in hypertrophied muscles. 85,86 These effects are arrhythmogenic and may accelerate progression of heart failure. 87 Angiotensin II also increases L-type calcium channel current and increases the expression of the L-type calcium channel α 1C subunit via a protein kinase C-dependent pathway. This effect is important in the remodeling process observed in atrial fibrillation (AF). 88 Animal models with cardiac-restricted ACE overexpression show marked atrial dilatation, atrial fibrosis, and atrial fibrillation. 89 Several clinical trials showed that ACE inhibitors and ARBs prevent the recurrence of AF, especially after cardioversion and in patients with LV dysfunction, lending more support to their antiarrhythmic effects. 90 –92
The ACE inhibitors decrease preload and afterload, which decreases myocardial oxygen demand and LV end-diastolic pressure. They also block angiotensin II production and inhibit the breakdown of bradykinin. 52 Blocking angiotensin II production prevents the progression of ventricular remodeling and reduces ventricular dilatation and fibrosis. 78 The ACE inhibitors result in the reduction in potassium depletion and have several effects on the autonomic nervous system via enhanced baroreflex sensitivity and hemodynamics, which can lead to reduced sympathetic and parasympathetic tone and circulating catecholamines. 78 Angiotensin II could persist despite treatment with ACE inhibitors, since it can be formulated by non-ACE-I-dependent pathways. The ARB can block AT1 receptors without an increase in bradykinin levels. 93 In the setting of acute MI, pretreatment with angiotensin receptor blockers prevents LV remodeling and blunts the increase in constitutive microtubulin in both the infarcted and the noninfarcted left ventricle. 94
Even with the utilization of ACE inhibitors or ARBs, there is no full suppression of aldosterone synthesis. Aldosterone promotes sodium retention, increases potassium secretion in the urine, and activates fibroblasts leading to myocardial and vascular fibrosis. 95 Myocardial fibrosis may increase the risk of ventricular arrhythmias by causing variations in the ventricular conduction times. Aldosterone receptor blockers prevent SCD by controlling potassium loss, blocking aldosterone effect on the formation of collagen, and increasing the myocardial uptake of norepinephrine, which decreases sympathetic activation. 93,96 Spironolactone decreases the level of serum markers of collagen synthesis at 6 months, which correlates with survival benefit. 96
Effect of ACE-I on SCD Prevention in Post-MI Patients and in Patients With Heart Failure
Three post-MI trials, SAVE, Trandolapril Cardiac Evaluation I (TRACE-I), and AIRE, specifically investigated the impact of ACE inhibitors on mortality and morbidity in post-MI patients who have LV dysfunction.
Survival and Ventricular Enlargement was a randomized, double-blind placebo controlled trial that evaluated the use of captopril (n = 1115) versus placebo (n = 1116) in post-MI patients with LVEF ≤40%. Randomization was done 3 to 16 days post-MI. During an average of 42 months, there was an 18% relative risk reduction in all-cause mortality with captopril compared to placebo. However, there was a nonsignificant trend toward lower SCD in patients taking captopril (HR 0.83, 95% CI 0.63-1.8). 97
Trandolapril Cardiac Evaluation I was designed to examine whether patients with a recent MI and LV dysfunction would benefit from long-term treatment with ACE inhibitors. A total of 1749 patients 3 to 7 days post-MI with echocardiographic evidence of LV dysfunction (EF ≤ 35%) were randomized to trandolapril (n = 876) or placebo (n = 873). During the follow–up, the relative risk of death from any cause in the trandolapril group versus the placebo group was 0.78 (95% CI 0.67-0.91). The trandolapril group also showed a significant reduction in sudden death versus the placebo group (HR 0.76, 95% CI 0.59-0.98, P = .03). 98 The TRACE-I was the first placebo-controlled trial to show a significant reduction in sudden death with the use of the ACE inhibitors.
The Acute Infarction Ramipril Efficacy trial once again looked at the use of ACE-I in the post-MI patient who had clinical or radiological evidence of CHF to receive ramipril (n = 1014) versus placebo (n = 992). After 15 months of follow-up, there was a 27% reduction in the risk of death with ramipril compared to placebo. In this study, ramipril also reduced the risk of sudden death by approximately 30% compared to placebo (P = .011). 99
A further meta-analysis looked at 15 trials including SAVE, TRACE-I, and AIRE to evaluate the effect of ACE-I on sudden death post-MI. This meta-analysis revealed a significant reduction in the risk of sudden death with an odds ratio of 0.80 (95% CI 0.70-0.92). 100
Currently, only 3 trials have reported the results of SCD in patients with systolic heart failure taking ACE-I. The Cooperative North Scandinavian Enalapril Survival (CONSENSUS) study was designed to evaluate the effect of enalapril compared to placebo on mortality in patients with severe systolic heart failure (class IV). This study randomized 253 patients to either enalapril (n = 127) or placebo (n = 126) in addition to conventional therapy. The CONSENSUS showed a 40% reduction in mortality after 6 months of treatment and a 27% reduction at the end of the study. The greatest reduction in mortality was in death caused by progression of pump failure. 101
The Studies of Left Ventricular Dysfunction (SOLVD)-Prevention trial was designed to determine whether an ACE inhibitor, enalapril, could reduce mortality, the incidence of heart failure, and the rate of hospitalizations in patients with EF ≤ 35%, with mild-to-moderate heart failure (class II or III). Following randomization, patients received double-blind treatment with either placebo (n = 1284) or enalapril (n = 1285). There was a 16% reduction in mortality due to progression of heart failure, but no clear reduction in SCD was noted. 102
The V-HeFT-II trial was the first trial to suggest the effect of ACE inhibitors on sudden death in patients with heart failure. This trial compared the effects of enalapril with hydralazine and isosorbide dinitrate on mortality in patients with NYHA class II-III. After randomization, double-blind treatment was instituted with enalapril (n = 403) versus hydralazine/isosorbide dinitrate (n = 401). Interestingly, the mortality curves of the treatment arms separate early after randomization. There was a 28% relative risk reduction with enalapril compared to hydralazine and isosorbide dinitrate (P = .016). The overall reduction in mortality associated with enalapril was due to a reduction in the incidence of sudden death. 103
Effect of ARBs on SCD Prevention in Patients With CHF
The ARBs were studied extensively and compared or added to ACE inhibitors in the treatment of patients with heart failure (both chronic systolic heart failure and post-MI LV dysfunction). The effects of ARB on mortality in randomized controlled trials in this patient population have been conflicting.
The Evaluation of Losartan in the Elderly (ELITE) study is the only ARB trial to demonstrate a reduction in sudden death. This prospective, double-blind, randomized, parallel group controlled clinical trial compared the safety and efficacy in the treatment of CHF with the use of losartan versus captopril. Patients were randomly assigned to losartan (n = 352) versus captopril (n = 370). Follow-up at 48 weeks showed a 45% reduction in all-cause mortality with a relative risk reduction of 36% in the incidence of SCD with losartan compared to captopril. 104
The Candesartan in Heart Failure: Assessment of Reduction in Mortality and Morbidity-Added (CHARM-ADDED) trial compared candesartan at a mean dose of 32 mg daily or placebo added to captopril in 2548 patients with NYHA class II to III heart failure and mean LVEF of 28%. This trial showed a 15% reduction in total mortality when adding candesartan to captopril, but it did not specifically study SCD. 105 Similarly, the Valsartan Heart Failure Trial (Val-HeFT) studied 5010 patients with NYHA class II to III heart failure and mean LVEF of 27%. The study compared valsartan at a dose of 160 mg orally twice daily and placebo added to ACE inhibitors. The study also showed no difference in all-cause mortality but showed a 13% relative risk reduction in cardiovascular morbidity and mortality drive by decreasing heart failure admission. The study did not specifically monitor SCD. 106
The Valsartan in Acute Myocardial Infraction (VALIANT) trial studied 14 703 patients with post-MI LV dysfunction, mean LVEF of 35%, and Killip class I to II heart failure and compared Valsartan 160 mg orally twice daily versus valsartan 80 mg orally twice daily added to captopril at 50 mg 3 times daily versus captopril at 50 mg orally 3 times daily. There was no difference between the treatment groups when it comes to all-cause mortality or cardiovascular mortality. 107
The ELITE II was designed to compare the effects of losartan and captopril on all-cause mortality and sudden death or resuscitated cardiac arrest. Similar to ELITE, patients were randomly assigned to losartan (n = 1578) or captopril (n = 1574). After 1.5 years of follow-up, there was no statistical difference in all-cause mortality, sudden death, or resuscitated cardiac arrest (losartan 9% versus captopril 7.3%, P = .08) between the 2 groups. 108 It is unclear why the results of ELITE I and ELITE II differed. This could be due to chance, to the smaller numbers, and shorter follow-up period in ELITE I compared to ELITE II. On the other hand, the failure of ELITE II to show superiority of losartan over captopril could be due to the low dose (50 mg) of losartan used. The HEAAL trial showed that a higher dose of losartan (150 mgs once daily) is superior to low-dose losartan (50 mg once daily) in decreasing mortality and hospitalization in patients with systolic heart failure. 109 It is also possible that these medications would have shown a reduction in overall mortality and sudden death when compared to placebo and not ACE-I, since both will block the effects of angiotensin II. The CHARM-alternative trial showed a reduction in cardiovascular mortality and hospitalization when using candesartan compared to placebo in patients intolerant to ACE-I. 110 Currently, there is no clear evidence that ARBs have a direct effect on SCD prevention, and they currently remain second line in patients with systolic heart failure after maximizing the treatment with ACE inhibitors or if the patient is intolerant to ACE inhibitors. 111
Effect of Aldosterone Antagonists on SCD Prevention in Post-MI Patients and in Patients With CHF
The Randomized Aldactone Evaluation Study (RALES) is a randomized, double-blind placebo controlled trial that compared spironolactone (n = 822) versus placebo (n = 841) in patients with severe heart failure. Patients enrolled had class III or IV heart failure and were being treated with an ACE inhibitor, loop diuretic, and had an EF ≤ 35%. This trial was ended prematurely when analysis found that spironolactone demonstrated a 31% reduction in cardiac death. This reduction was due to a 36% in death related to progressive heart failure and a 29% reduction in SCD. 112
The Eplerenone Post Myocardial Heart Failure Efficacy and Survival Study (EPHESUS) was conducted to evaluate the effect of aldosterone blocker, eplerenone on morbidity, and mortality among patients with acute MI complicated by LV dysfunction and heart failure. In this double-blind, placebo-controlled study, patients were randomly assigned to eplerenone (n = 3313) versus placebo (n = 3319) in addition to optimal medical therapy. eplerenone demonstrated a reduction in death from cardiovascular causes or hospitalization for cardiovascular events (relative risk 0.83; 95% CI 0.72-0.94; P = .005). There was also a reduction in sudden death from cardiac causes (relative risk 0.79; 95% CI 0.64-0.97; P = .03). 113
Although there is a clear benefit of aldosterone blockers in the setting of heart failure and post-MI patients., this does not come without a price. Most of the patients with heart failure will have renal impairment, and if care is not taken to follow the kidney function and the potassium levels, hyperkalemia and arrhythmic death could occur obviating the benefit of these medications. In the Atrial Fibrillation and Congestive Heart Failure (AF-CHF) trial, there was a 1.4 increase in total mortality and cardiovascular mortality, driven mainly by an increase in SCD (HR 2.0, 95% CI 1.3-3.0, P = .001). 114 There was a high prevalence of renal dysfunction in this study, and 45% of the patients were on aldosterone blockers, mainly spironolactone. This was a post hoc analysis of the trial, and care should be taken in extrapolating the data. Although the randomized studies show a clear benefit of aldosterone blockers in decreasing all-cause mortality and SCD, care should be taken to follow the kidney function and monitor potassium levels.
Renin–Angiotensin–Aldosterone System Inhibitors/Blockers in Patients With Heart Failure and Normal EF
The presence of myocyte hypertrophy and collagen deposition in patients with HFNEF justifies the use of renin–angiotensin–aldosterone inhibitors/blockers from a mechanistic stand point. In animal models, treatment with captopril leads to regression of LV hypertrophy and reversal of proarrhythmic properties of the myocardium. 71 The losartan Intervention For Endpoint reduction in Hypertension (LIFE) study showed that losartan leads to regression of LV hypertrophy on the electrocardiogram, which was associated with reduction in SCD. 115
The Perindopril in elderly people with chronic heart failure (PEP-CHF) trial was the first trial to examine the use of ACE-I in patients with HFNEF. Patients older than 70 years of age were enrolled if they had LVEF >40%, evidence of LV hypertrophy, and abnormal filling pressures on mitral inflow velocity Doppler as well as a diagnosis of HF. There was no difference in mortality in heart failure or hospitalizations. The trial has the same shortcomings the SENIOR trial had in its design, since the accurate definition of HFNEF had to satisfy 3 of 9 criteria, one of which is MI. As a result, 40% of the patients had a history of coronary artery disease and eccentric hypertrophy as opposed to concentric hypertrophy. 116
The CHARM-preserved trial compared candsartan to placebo in patients with HF and normal LVEF. However, there was no significant effect of candesartan on cardiovascular mortality. 117 The CHARM preserved also enrolled may patients who had history of MI (45% of the patients enrolled had history of prior MI and coronary artery disease). This confounds the results, since these patients were found to have eccentric hypertrophy. The Candesartan in Heart Failure Reduction in Mortality Echocardiographic Substudy (CHARMES) 118 showed that 66% of the patients had LVMI of 111 g/m2, which is in the normal range. 119
Finally, the Irbesartan in Heart Failure with Preserved Systolic Function (I-PRESERVE) trial randomized 4128 patients to irbesartan versus placebo. Most of the patients had normal LVEF. Only 24% of these patients enrolled had history of coronary artery disease. There was no difference in mortality or hospitalization in patients taking irbesartan compared to placebo. 120
Overall, patients enrolled in the above trials were mostly selected based on the presence of normal LVEF and heart failure symptoms, but the cutoff of LVEF varied and the presence of diastolic dysfunction was mostly based on mitral inflow velocity that changes with age and other conditions. The high prevalence of coronary artery disease was found to be responsible for enrolling patients with eccentric as opposed to concentric hypertrophy and remodeling. The new ESC guidelines for diagnosing HFNEF offer a better definition of HFNEF that was not present during the design of these trials. 68 This offers huge opportunities for further clinical studies in this patient population.
Statins (3-Hydroxy-3-Methylglutaryl Coenzyme-A Reductase Inhibitors) and SCD Prevention
Potential Mechanisms of 3 Hydroxy-3-Methylglutaryl Coenzyme A Reductase Inhibitors on SCD Prevention
Statins (3-hydroxy-3-methylglutaryl coenzyme-A reductase inhibitors) have been shown to decrease cardiovascular morbidity and mortality in both primary and secondary prevention trials.121–123 Statins are known to stabilize the plaque and to even promote plaque regression. 124 This stabilization improves myocardial perfusion and oxidative stress and reduces the risk of plaque rupture. 125 This leads to decreased ischemic events and arrhythmic events, since even small areas of ischemia can promote reentry, induce ventricular arrhythmias, and lead to SCD. Statins improve endothelial function by increasing nitric oxide production from endothelial cells, and they reduce ischemia-mediated oxidative stress and intracellular calcium overload. 126,127 They also have anti-inflammatory actions, reduce C-reactive protein, and decrease endothelin-1 secretion. 128 All these effects will decrease myocardial ischemia, limit myocardial injury, and prevent myocyte hypertrophy. 129,130 Statins have been shown to improve LVEF in patients with dilated cardiomyopathy even after short duration of therapy. 131,132 Pravastatin was shown to increase contractile function of hibernating myocardium in a canine model by increasing myocyte cell proliferation and reentry to the growth phase of the cardiac cell cycle. 133 Furthermore, statins also decrease QT dispersion, improve autonomic tone and heart rate variability, and modulate ion channels independent of their lipid-lowering effects. 134,135 These “pleiotropic” properties may account for their effect on reducing arrhythmic death in both ischemic and nonischemic cardiomyopathy.
Effect of Statin Therapy on Shock Burden and SCD in Post-MI Patients and in Patients With CHF
Statins are widely accepted as preventing coronary heart disease death and MI; however, their effect on SCD prevention is unclear. Randomized Trials in post-MI patients showed the benefits of statins on overall mortality but failed to show benefit on SCD prevention.121,136,137 However, observational data from hospitalized patients with MI showed that early statin administration (within 24 hours) of an acute MI led to a decrease in the incidence of VT/VF. 138 Statins appear to decrease appropriate shocks in patients who have ICDs whether or not they received them for primary or secondary prevention of SCD. In a subanalysis of AVID trial, a secondary prevention trial that compared antiarrhythmic drugs to ICDs in patients who survived a cardiac arrest, patients who received statins had a lower risk of ventricular arrhythmias compared to those who are not on statins. 139 This was also demonstrated in the MADIT-II. Post hoc analysis of MADIT-II showed that patients receiving statin therapy >90% of the time had a significantly reduced cumulative rate of ICD therapy for VT/VF or cardiac death. 140 Subsequently, an analysis of SCD-HeFT trial data was undertaken to evaluate the impact of statin use in heart failure. The SCD-HeFT studied 2521 patients with functional class II and III heart failure having LVEFs ≤35%. The cause of CHF was ischemic in 52% of the study patients. Statin use was reported in 965 (38%) of 2521 patients at baseline and 1187 (47%) at last follow-up with the median time to follow-up of 45.5 months. This analysis revealed that mortality reduction related to statin therapy (HR = 0.70, 95% CI 0.58-0.83) was identical in both ischemic and nonischemic cardiomyopathy (HR 0.69 vs 0.67, respectively). 141
Statins decrease the risk of arrhythmic death in patients with nonischemic cardiomyopathy. The Defibrillators in Non-Ischemic Cardiomyopathy Treatment Evaluation (DEFINITE) trial enrolled 458 patients with nonischemic cardiomyopathy to receive a defibrillator versus optimal medical therapy. More than 90% of the patients were taking β-blockers and ACE inhibitors. Statin therapy was not randomized in this trial. There was a 78% relative risk reduction of death in patients taking statins (n = 110) versus those not on statins (n = 348). 142 The same observation was seen in MADIT with cardiac resynchronization therapy (MADIT-CRT) trial. Of the 821 with nonischemic cardiomyopathy enrolled in MADIT-CRT, there were 499 patients taking statins. Multivariate analysis showed that time-dependent statin therapy was independently associated with a 77% reduction in the risk of VT/VF or death (P < .001) and with a 46% reduction in the risk of appropriate implantable cardioverter defibrillator shocks (P = .01) regardless if the patient received a cardiac resynchronization device or if they were enrolled to receive a defibrillator only. 143
Two major clinical trials on the use of rosuvastatin in patients with systolic heart failure failed to show mortality benefit. The first is the Controlled Rosuvastatin Multinational Trial in Heart Failure (CORONA) trial that studied 5011 patients older than 60 years of age with NYHA class II, III, and IV ischemic heart failure. Patients were randomized to receive 10 mg of Rosuvastatin versus placebo. Despite 45% reduction in low-density lipoprotein cholesterol and 37% reduction in high-sensitivity C-reactive protein, there was no difference in the primary outcome of cardiovascular death or death from any cause. Treatment with rosuvastatin led to a decrease in hospitalization and was well tolerated by this population of patients. 144 The lack of benefit could be due to chance, short follow–up, or age of the population studied. The GISSI-HF trial randomized patients with NYHA class II-IV heart failure irrespective of cause or LVEF to 10 mg of rosuvastatin versus placebo. There was no difference in mortality in the patients assigned to rosuvastatin (2285 patients) compared to patients assigned to placebo (2289 patients) after 4 years of follow-up (HR 1.00, 95% CI of 0.89-1.12, P = .94). There was also no difference in hospitalization between the 2 groups of patients. 145
Why did the post hoc analysis from MADIT-II, SCD-HeFT and MADIT-CRT differ from the results of CORONA and GISSI-HF trials? One possibility is related to the limitations of post hoc analysis and possibility of bias and chance. Another is related to the type of statin used by the patients. Although CORONA and GISSI-HF studied rosuvastatin, patients in MADIT-CRT and SCD-HeFT were taking different types of statins at different doses. Furthermore, patients in MADIT-CRT had mild heart failure (NYHA class I and II) compared to the majority of patients in CORONA and GISSI-HF who had moderate to severe HF (mostly classes III and IV). Whether different statins have different effects in patients with heart failure needs to be further clarified. Atorvastatin and simvastatin were shown to improve LVEF in patients with dilated cardiomyopathy. 82,83 Finally, pravastatin was shown in an animal model to increase contractile performance of hibernating myocardium by increasing myocyte cell cycle reentry and proliferation. 133 It has been shown that β-blockers are more effective in patients who have more viable myocardium and are less effective when the scar is >75% of wall thickness. 37 Whether this is also true in patients taking statins is another area that needs to be explored. Taking all the available data together, statins appear to reduce the frequency of ICD shocks even in patients taking β-blockers and ACE-I. They appear to be useful in patients with systolic heart failure who have received a defibrillator for primary or secondary prevention and are generally well tolerated. More randomized trials utilizing different statins at different doses are needed to clarify the effects of statins on sudden death in patient with mild, moderate, and severe heart failure.
Conclusions and Future Directions
The SCD remains a challenge for health providers and policy makers. Whether more stringent guidelines for prevention and screening will be applied is balanced by the enormous costs, this will place on the health care system. In order to identify the groups at risk of SCD, there must first be a standardization of the definition. The worldly variation in the definition of SCD of 1 hour from onset of symptoms to 24 hours not only affects epidemiological data but also alters clinical trial outcomes when evaluating the effectiveness of treatment and prevention options. 6
Currently, antiarrhythmic medications have failed to show any benefit of SCD prevention, although traditional heart failure medications have been shown to decrease total mortality, SCD, and defibrillator shocks. They are used only in a small subset of patients at risk of SCD, since most of the patients who have SCD have it as a first presentation and do not have systolic heart failure. 3,4 A focus on coronary artery disease prevention is warranted given the fact that coronary artery disease is present in >50% of patients older than 35 years of age who died suddenly and underwent autopsy. 3 But even screening and prevention carries challenges. Intravascular ultrasound studies in hearts of cardiac transplant donors showed significant atherosclerotic plaque even in individuals younger than 20 years. 146 For primary prevention of coronary artery disease, most of the primary prevention algorithms will not initiate statin therapy till the patient is in his or her 50s, even if he or she has multiple risk factors. 147 The cost of starting this treatment is also enormous, especially if it is started on a global scale at a young age, and it is not without side effects. Genetic studies to identify patients at risk of SCD are important and are still under development. Well-designed clinical trials that target patients with heart failure and normal EF are needed to treat this large population of patients that are at risk. Identifying other parameters that can help risk stratify the patients at risk of sudden death other than the LVEF is another area that needs further studies. Preventing SCD is definitely a challenge for the 21st century clinician and might remain so for the near future.
Footnotes
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.