
Abstract
Purpose:
Fibroblast activity promotes adverse left ventricular (LV) remodeling that underlies the development of ischemic cardiomyopathy. Transforming growth factor-β (TGF-β) is a potent stimulus for fibrosis, and the extracellular signal-regulated kinases(ERK) 1/2 pathway also contributes to the fibrotic response. The thrombin receptor, protease-activated receptor 1 (PAR1), has been shown to play an important role in the excessive fibrosis in different tissues. The aim of this study was to investigate the influence of a PAR1 inhibitor, SCH79797, on cardiac fibrosis, tissue stiffness and postinfarction remodeling, and effects of PAR1 inhibition on thrombin-induced TGF-β and (ERK) 1/2 activities in cardiac fibroblasts.
Methods:
We used a rat model of myocardial ischemia–reperfusion injury, isolated cardiac fibroblasts, and 3-dimensional (3D) cardiac tissue models fabricated to ascertain the contribution of PAR1 activation on cardiac fibrosis and LV remodeling.
Results:
The PAR1 inhibitor attenuated LV dilation and improved LV systolic function of the reperfused myocardium at 28 days. This improvement was associated with a nonsignificant decrease in scar size (%LV) from 23 ± % in the control group (n = 10) to 16% ± 5.5% in the treated group (n = 9; P = .052). In the short term, the PAR1 inhibitor did not rescue infarct size or LV systolic function after 3 days. The PAR1 inhibition abolished thrombin-mediated ERK1/2 phosphorylation, TGF-β and type I procollagen production, matrix metalloproteinase-2/9 activation, myofibroblasts transformation in vitro, and abrogated the remodeling of 3D tissues induced by chronic thrombin treatment.
Conclusion:
These studies suggest PAR1 inhibition initiated after ischemic injury attenuates adverse LV remodeling through late-stage antifibrotic events.
Keywords
Introduction
Mortality due to myocardial infarction (MI) continues to decline, while the incidence of heart failure post-MI has remained fairly consistent. 1 The number of heart failure hospitalizations remains unchanged, 2 and the long-term prognosis for patients with acute heart failure remains poor. 3 Cardiac fibrosis is now widely recognized as part of the pathogenesis of post-MI remodeling and heart failure. 4 After MI, the infarct border zone is infiltrated by neutrophils and macrophages. Cardiac fibroblasts transform into myofibroblasts and migrate into the border zone, where they promote contraction of the scar. Myofibroblasts, detected by the presence of α smooth muscle actin (α-SMA), represent the main cell type involved in the development of fibrosis by increasing the production of transforming growth factor-β (TGF-β), 5,6 expression of extracellular matrix metalloproteinases (MMPs), enhanced collagen deposition, and contraction of the surrounding matrix. 7
Protease-activated receptor 1 (PAR1) has been described as the most highly expressed G-protein-coupled receptor in adult rat cardiac fibroblasts. 8 Activation of PAR1 on neonatal cardiac fibroblasts leads to transient increase in intracellular calcium and extracellular signal-regulated kinase (ERK)1/2 phosphorylation, increased DNA synthesis, and cell proliferation. 9 The ERK1/2 signaling has been identified as a critical pathway in PAR1-mediated profibrotic activities in lung fibrosis 10 ; however, PAR1-induced fibrotic activities in the heart have not been previously explored.
The PAR1 has been implicated in acute and chronic cardiac injury. The PAR1 is upregulated and activated in patients with ischemic cardiomyopathy. 11,12 We have previously shown that PAR1 inhibition prior to myocardial ischemia–reperfusion (IR) decreases infarct size acutely in rats. 13 In contrast, PAR1-deficient mice subjected to IR were not protected from acute myocardial injury, yet these mice were found to have reduced left ventricular (LV) dilation and preserved cardiac function after 2 weeks. 14 Cardiac-specific overexpression of PAR1 induced cardiac hypertrophy in mice. 14 Likewise, Jaffre et al more recently showed that upregulation of MMP-13 in a nonischemic heart failure model promotes cardiac hypertrophy through PAR1. 15 Although these studies clearly implicate that injury-induced PAR1 activation leads to cardiac injury and hypertrophy, the contribution of PAR1 activation in cardiac fibrosis and in cardiac fibroblast activities has not been investigated previously.
We hypothesized that the ERK1/2 pathway was responsible for PAR1-mediated remodeling activities of cardiac fibroblasts including myofibroblast transformation, collagen synthesis, and matrix remodeling. Therefore, we sought to determine whether chronic treatment with a PAR1 inhibitor after IR injury attenuates myocardial fibrosis and improves LV remodeling activities to maintain cardiac function. To analyze PAR1-mediated profibrotic activities in fibroblasts, we used isolated cells and 3-dimensional (3D) tissue models to determine the effects of PAR1 on cellular contractility and tissue remodeling.
Methods and Materials
The models and measurements used for this study are shown in Table 1.
Models and Measurements Used for This Study.

Abbreviations: IR, ischemia-reperfusion; LV, left ventricular; TGF, transforming growth factor-β; 3D, three dimensional.
Rat Model of Myocardial IR Injury and LV Remodeling
Male Sprague-Dawley (SD) rats at 8 weeks of age (250-300 g) used in this study received humane care in compliance with the Guide for the Care and Use of Laboratory Animals formulated by the National Research Council (1996). The protocol was approved by the Institutional Animal Care and Use Committee at the Medical College of Wisconsin.
To determine whether PAR1 inhibition would limit LV remodeling in vivo, we used a rat model of ischemia caused by 30 minutes of left coronary artery (LCA) occlusion followed by reperfusion. Transient LCA occlusion was performed for the determination of the area at risk (AAR) and infarct size described previously. 16 In the acute IR study, the rats were divided into 2 groups (IR + placebo and IR + PAR1 inhibitor; n = 6/group) and underwent 30-minute LCA occlusion followed by 120 minutes of reperfusion. The PAR1 inhibitor, SCH79797 (25 µg/kg) was injected 15 seconds after the onset of reperfusion. For each chronic study (3 days and 28 days), the animals were randomly assigned to 4 groups (n = 12/group; 1) Sham-operated + placebo (Sham), (2) Sham-operated + PAR1 inhibitor (Sham + PAR Inh), (3) IR + placebo (IR), and (4) IR + PAR1 inhibitor (IR + PAR Inh). Rats in the treated groups received a bolus dose of the SCH79797 (25 µg/kg; intraperitoneally [ip]) 15 seconds after the onset of reperfusion followed with a continuous SCH79797 infusion (25 µg/kg/d; ip) via osmotic pumps (Alzet, Cupertino, California) for 3 or 28 days. Sham and IR rats received an equivalent volume of sterile saline as placebo. Sham-operated rats did not undergo coronary artery occlusion.
Systolic blood pressure (BP) and heart rate were measured by a computerized tail-cuff system (Visitech Systems, Apex, North Carolina), as described previously. 17 Extracted lungs were weighed before and after drying at 90°C for 48 hours. The dry to wet ratio was calculated as an index for pulmonary edema. 18 Six hearts from each group were harvested by the end of the observation period for histology and infarct size measurements; the remaining hearts were used for biochemical and molecular assays.
Infarct Size and Histological Analysis
At the end of 120-minute reperfusion, the coronary artery was reoccluded, and the AAR zone was delineated by intravenous injection of 0.5% Evans blue. Hearts were sectioned and incubated in 1% triphenyltetrazolium chloride in phosphate buffer (pH 7.4, 37°C) for 15 minutes to define white necrotic tissue when fixed in 10% formalin for 24 hours. Infarct size, AAR, and LV area were determined by computerized planimetry using J-Image 1.45-s software (NIH, Bethesda, Maryland). In the acute studies, AAR was consistent between the groups (55.3 ± 2.77 as a % LV).
To visualize cardiac infarct and scarring in the chronic studies, heart sections were prepared as described previously 19 and stained with hematoxylin and eosin or picrosirius red. The infarcted area to LV ratio was determined by planimetry as above. At 28 days, the collagen content in the scarred areas was determined using MetaVue Imaging System (Version 5.0r1, 2002) color threshold function. 20 The infarct or scar area was then calculated as a percentage of the total LV area. The border zone was defined as the total myocardial area containing the scar. Immunohistochemical staining was performed using an anti-α-SMA antibody (A2547, Sigma, St Louis, Missouri). 21 Quantification of myofibroblast density in the border zone was performed using MetaVue. Three random images of the infarct border zone (×20 magnification) were selected and thresholded for calculation of the percentage of the stained area. 22
Echocardiography
Transthoracic echocardiography was performed in anesthetized (2% isoflurane) animals at baseline and during reperfusion (at 3 days and 28 days). Measurements and data analyses were performed by an investigator (L.M.H.) blinded to the study groups. Animals were studied in the left lateral decubitus position with a commercially available echocardiographic system (Vivid 7, General Electric, with an 11-MHz M12-L linear array transducer, GE Healthcare, Waukesha, WI), as previously described. 23 From the cardiac short axis at papillary level, an M-mode trace of the LV was obtained; left ventricular end diastole diameter and systole were measured. The LV ejection fraction (EF) was measured by Simpson method from 2-dimensional parasternal long- and short-axis (basal, mid, and apical level) views, as described previously. 24 The LV mass was derived from the anteroseptal thickness (AST) and inferolateral thickness (ILT) using the formula: 0.8 (1.04[(ILT + LVIDd + AST)3 − LVIDd3]) + 0.6. 24 Three consecutive heart beats were measured and averaged.
Cell and Tissue Culture
Adult rat cardiac fibroblasts were prepared and cultured as described previously. 25 Briefly, the left ventricles were isolated from excised rat hearts, minced, and digested in 0.1% collagenase type II (Gibco, Grand Island, New York) and 0.25% trypsin. The cells were plated in Dulbecco modified Eagle medium (DMEM) supplemented with 10% fetal bovine serum (FBS, Gibco) and antibiotics (penicillin and streptomycin), and after 60 minutes the unattached or weakly attached cells were removed.
To extrapolate changes in mechanical properties of myocardium initiated by cardiac fibroblast activities, 3D-engineered tissues were fabricated by embedding the isolated cardiac fibroblasts in collagen hydrogels as previously described 26 (Figure 1A). Briefly, the cells were mixed with neutralized rat tail collagen (1 mg/mL; Millipore, Billerica, Massachusetts) and DMEM containing 10% FBS. A small amount (250 µL) of this solution was poured into each well of an 8-well polycarbonate mold and incubated at 37°C for 30 minutes to allow the cells to become trapped within the polymerized collagen matrix. The tissues were then cultured in DMEM supplemented with 10% FBS and antibiotics at 37°C for 2 to 3 days. During this time, the entrapped cells (1 million cells/1 mL gel solution) remodel and compact the tissue by cell traction force. Final concentrations of cells and collagen in the remodeled samples are ∼18 million/mL and ∼18 mg/mL, respectively. For mechanical measurements, culture medium was switched to a HEPES-buffered DMEM, pH 7.4. All cardiac fibroblast tissues were prepared identically and tested following 2 to 3 days of incubation. Therefore, the size of each cardiac fibroblast tissue including its cross-sectional area was almost identical. The stiffness of the 3D-engineered tissue was measured by a probe connected to isometric force transducer using a device described previously 26 (Figure 1B). Resistance force is recorded as the probe stretched the tissue, and through biomechanical analysis tissue stiffness and cell force is determined (Figure 1C).
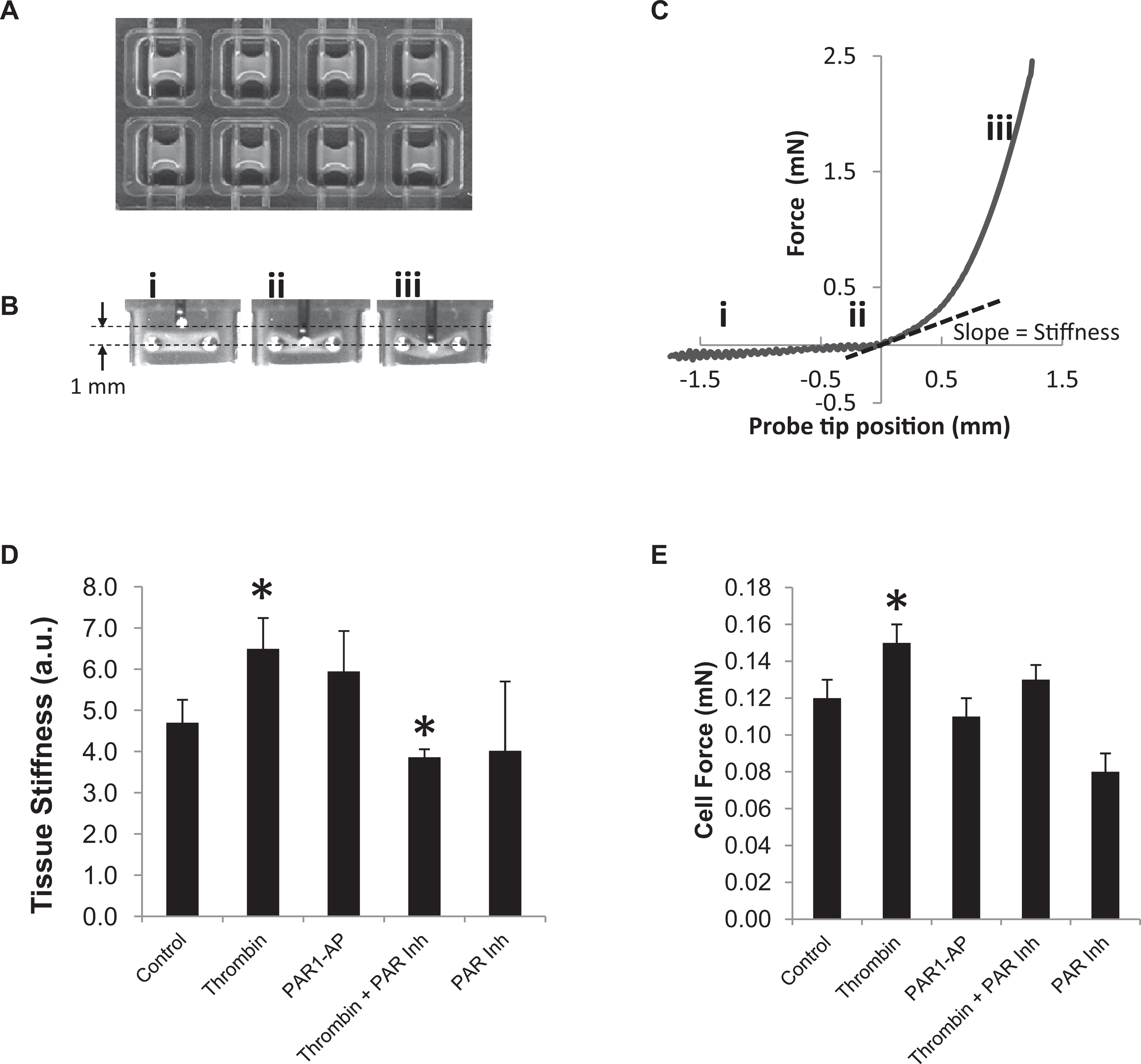
Thrombin-stimulated 3-dimensional (3D) cardiac fibroblast tissue models. A, 3D cardiac fibroblast tissue models were fabricated and cultured for 2 to 3 days prior to treatments in an 8-well chamber. B, A horizontal bar perpendicularly attached to a vertical force probe pushed at its mid-section and stretched 3D tissue gradually. C, The force resisted to stretching tissue pushed against the force probe was recorded through an isometric force transducer attached to the force probe. A mathematical simulation of stretching membrane was used to obtain tissue stiffness and cell force. D, Thrombin increased tissue stiffness over control values in 3D tissues, and protease-activated receptor 1 (PAR1) inhibition abrogated this response. E, Estimated cell force was also increased by thrombin treatment, but PAR1 inhibition did not attenuate significantly. Data in (D) and (E) are mean ± standard error of the mean (SEM; n = 15/group). *P < .05 versus control. AP indicates PAR1 activating peptide.
Treatment of Rat Cardiac Fibroblasts
Cardiac fibroblasts from third to fifth passages were seeded and grown to 70% confluence in 60-mm plates (for protein analysis) and 96-well plates (for collagen synthesis assays). After serum starving, the cells were treated with thrombin (10 U/mL) or a PAR1-activating peptide (TFLLR-amide, 100 μmol/L; Bachem, Torrance, California) with or without inhibitors (PD98059, 20 μmol/L or SCH79797, 1 μmol/L) for either 5 minutes (for Western blot analysis) or 24 hours (for enzyme-linked immunosorbent assay and zymography). Each condition was performed in triplicate and repeated in 3 independent experiments.
Immunofluorescence and Confocal Microscopy
Cardiac fibroblasts freshly isolated from SD adult rats were plated onto 4-well chamber slides (Thermo Fisher Scientific, Rochester, New York). After overnight starvation, thrombin (5 U/mL) with or without inhibitors were added for 24 hours. The cells were fixed and immunostained with a Cy3 labeled anti-α-SMA antibody (1:500 dilution). The nuclei were stained with 4',6-diamidino-2-phenylindole (DAPI; 1:10 000 dilution). A Nikon A1 confocal microscope was used to image α-SMA and DAPI staining (Nikon Instruments, Melville, New York).
Immunoblot Analysis
Protein extraction and immunoblot analysis was performed, as described previously. 13 Cells were directly lysed in buffer and cleared by centrifugation. Protein concentration was measured by the bicinchoninic acid protein method. Equal amounts of protein were run on a 10% polyacrylamide sodium dodecyl sulfate gel and transferred to a nitrocellulose membrane. After the membrane was blocked with 5% bovine serum albumin (BSA), it was incubated with a 1:1000 dilution of the primary antibody (phospho-p44/42 mitogen-activated protein kinase (Thr202/Tyr204) or glyceraldehyde 3-phosphate dehydrogenase antibodies from Cell Signaling Technology (Danvers, Massachusetts) or Abcam (Cambridge, Massachusetts). After washing, the blots were incubated with a secondary antibody, followed by enhanced chemiluminescence solution (Amersham Life Science, Arlington Heights, Illinois) and exposed to film for 1 minute.
Quantification of TGF
The TGF-β1 levels were determined using a TGF-β1 enzyme-linked immunoassay kit from R&D Systems (Minneapolis, Minnesota), according to the manufacturer’s instructions.
Quantification of Procollagen Type I
After treatment for 24 hours, the cells were fixed with 10% buffered formalin, washed, permeabilized with triton X-100 (0.3% in phosphate-buffered saline), and blocked with 2.5% BSA in phosphate-buffered saline Tween 20 (PBST). After washing, rabbit anti-rat collagen type I polyclonal antibody (1:500; Chemicon, Temecula, California) diluted in PBST with 1% BSA was applied for 1 hour at 37°C. The plate was washed and treated with a goat anti-rabbit Immunoglobulin G secondary antibody conjugated with horseradish peroxidase diluted in PBST with 1% BSA for 1 hour at 37°C. A substrate solution was made with Sigma FAST ophenylenediaminedihydrochloride (OPD) tablets dissolved in 20 mL of distilled water. Final concentrations are 0.4 mg/mL OPD, 0.4 mg/mL urea hydrogen peroxide, and 0.05 mol/L phosphate citrate, pH 5.0. The enzyme reaction was stopped after 30 minutes incubation at RT in the dark by the addition of 50 μL of 2 mol/L H2SO4 to each well. The plates were read at 490 nm.
Chemicals and Reagents
All reagents were purchased from Sigma (St. Louis, Missouri) unless stated otherwise. The specific PAR1 inhibitor, SCH79797 (N3-cyclopropyl-7-{[4-(1-methylethyl) phenyl] methyl}-7H-pyrrolo [3, 2-f] quinazoline-1, 3-diaminedihydrochloride) 27 was purchased from Tocris Bioscience (Ellisville, Missouri).
Statistical Analysis
All values are expressed as mean ± standard error of the mean unless otherwise stated. For variables for which only 2 groups existed, (ie, infarct size), a student t test was performed. A 1-way analysis of variance (ANOVA) followed by Dunnet post hoc test was used to analyze group differences for TGF-β, procollagen, and MMP activity. Echocardiographic and hemodynamic data across time were analyzed by 1-way repeated measures ANOVA followed by post hoc analysis with Dunnett test for multiple comparisons against baseline measurements or by 1-way ANOVA followed by post hoc analysis with Dunnett test for multiple comparisons against the Sham + placebo measurements (SigmaPlot; Systat Software, Inc, Richmond, California). A value of P < .05 was considered statistically significant.
Results
Animal Characteristics and Hemodynamics
For the chronic study, a total of 48 rats were initially included. In all, 1 rat in the Sham group and 3 rats in the IR + PAR Inh group died prior to recovery from anesthesia. Two rats in the IR group died within the first week and were excluded from the study. The final 42 rats were analyzed by echocardiography at the 3- and 28-day time points. Myocardial infarcts were confirmed by echocardiography at day 3, which revealed anteroseptal, anterior, and anterolateral wall akinesis and corresponding echodense regions (Figure 2

Representative 2-dimensional (2D) echocardiography images of the study groups. The parasternal short-axis view of the left ventricle at end diastole is shown at the baseline, 3 days, and 28 days. Representative images from the Sham group (rat #14), the ischemia-reperfusion (IR) + placebo group (rat # 44) and the IR + protease-activated receptor (PAR) inhibitor group (rat #3 and #19). The arrows point to the infarcted regions seen at 3 days. The infarcted regions are echodense and have corresponding wall motion abnormalities. The anterior and anterolateral wall akinesis is best represented by a flattening of those segments during end diastole.
The baseline characteristics of the rats in each group are summarized in Table 2. No significant differences between body weights, heart rate, or mean BP were found at baseline. Body weights increased in all the groups throughout the 28 days; however, there was a 6% increase in body weights of the IR group when compared to the Sham group. The PAR1 inhibitor did not decrease body weights significantly after IR. In addition, the heart rate decreased almost by half in the IR group at 28 days when compared to the baseline. Heart rate was maintained close to baseline in the Sham and IR + PAR Inh groups. The IR and IR + PAR Inh group had a small but significant increase in mean BP at 3 and 28 days when compared to baseline. The mean BP in the IR group also had a slight but still significant increase over the IR + PAR Inh group at 28 days.
Tissue Weights and Hemodynamic Parameters.

Abbreviations: BPM, beats per minute; d, day; Inh, inhibitor; IR, ischemia-reperfusion; nd, not defined; PAR, protease-activated receptor.
a P < .05 versus Sham + placebo.
b P < .05 vs. baseline.
c P < .05 versus IR + placebo
Compared to the Sham groups, the heart to body weight ratios increased by 18% in the IR groups. Chronic PAR1 inhibition did not significantly reduce this increase after IR. The PAR1 inhibition in the sham group also significantly increased the heart to body weight ratios (Table 2).
Chronic PAR1 Inhibition Decreases Cardiac Remodeling and Preserves Function In Vivo
Chronic PAR1 inhibition normalized the increase in systolic and diastolic dimensions and volumes (Figures 2 and 3A-E
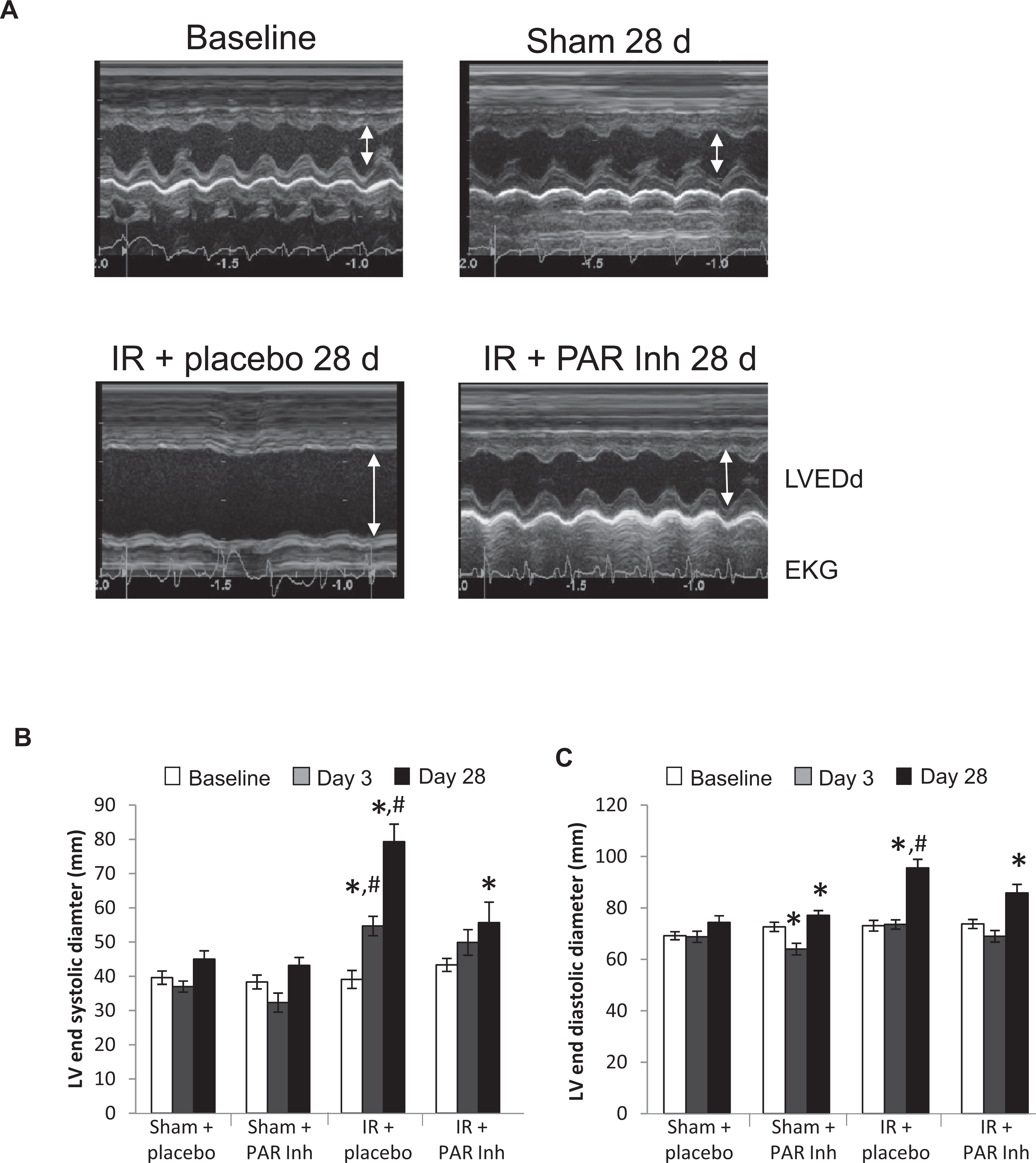
Time-dependent changes of left ventricular (LV) parameters. A, Representative M-mode echocardiograms obtained at baseline and at 28 days in the Sham group. All other groups were subject to 30 minutes ischemia followed by 28 days reperfusion with placebo (ischemia-reperfusion [IR] + placebo) or with the protease-activated receptor (PAR) 1 inhibitor (IR + PAR Inh). Time-dependent changes of (B) LV end-systolic diameter (C) LV end-diastolic diameter (D) LV end-systolic volume (E) LV end-diastolic volume (F) LV ejection fraction. Data are mean + standard error of the mean (SEM), (Sham + placebo, n = 12; Sham + PAR Inh, n = 11; IR + placebo, n = 10; IR + PAR Inh, n = 9). *P < .05 versus baseline. #P < .05 versus Sham + placebo. LVEDd indicates left ventricular end-diastolic diameter.
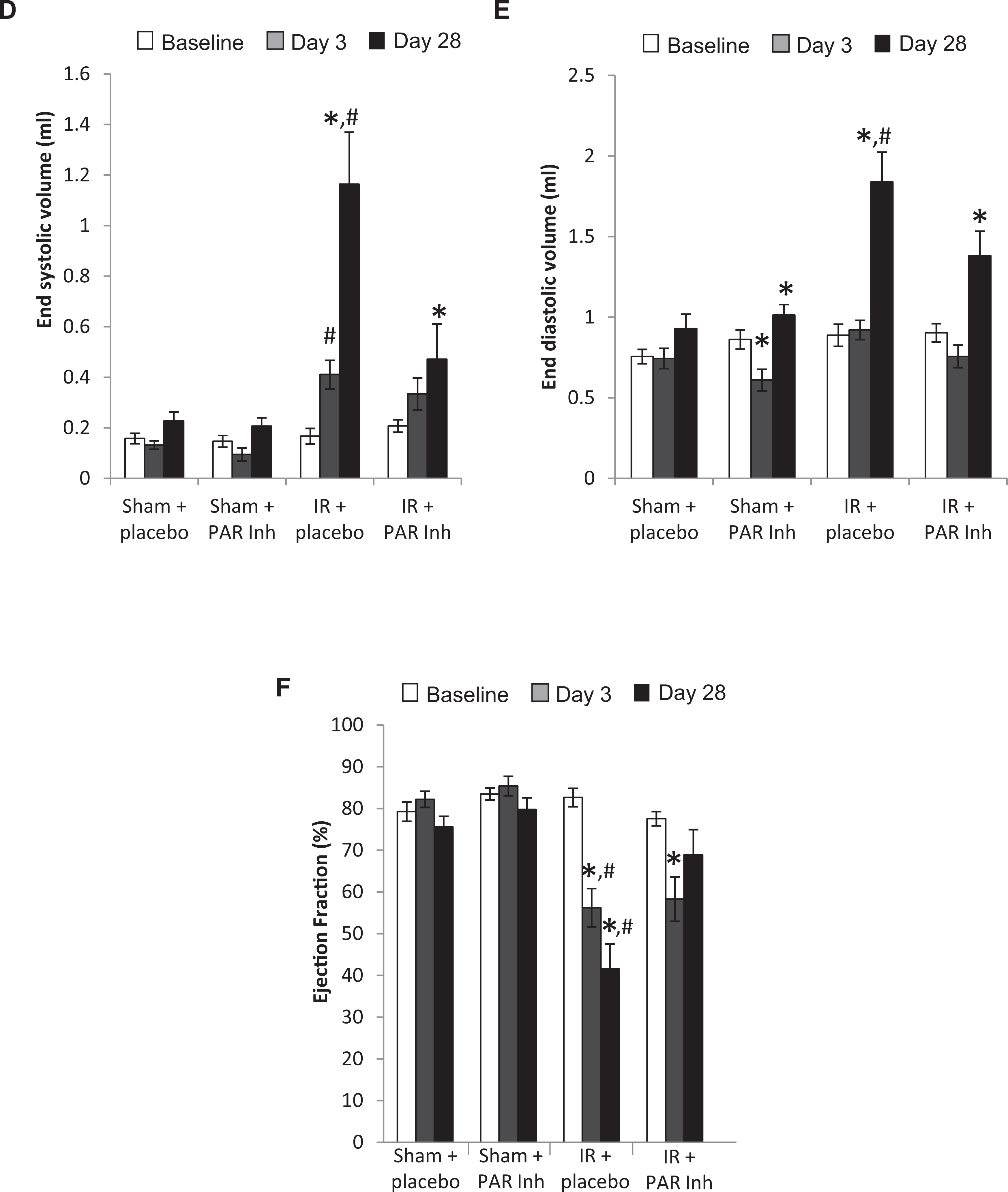
(Continued)
Chronic PAR1 inhibition over 28 days following the 30 minutes of ischemia showed a reduction in scar size (16% ± 5.5% LV vs 23% ± 4.1% LV control, P = .052) as determined by picrosirius red staining (Figure 4A and B). No increase in reactive fibrosis was detected in the noninfarcted region. The area of the border zone plus scar was not significant between the groups (Figure 4C). The dry to wet lung weight ratio, a marker of pulmonary edema, increased almost 2-fold in the IR versus Sham groups. This increase was completely abolished with PAR1 inhibition (Figure 4C). Figure 4D shows a representative of an IR lung compared to a representative lung from the Sham and IR + PAR Inh groups.
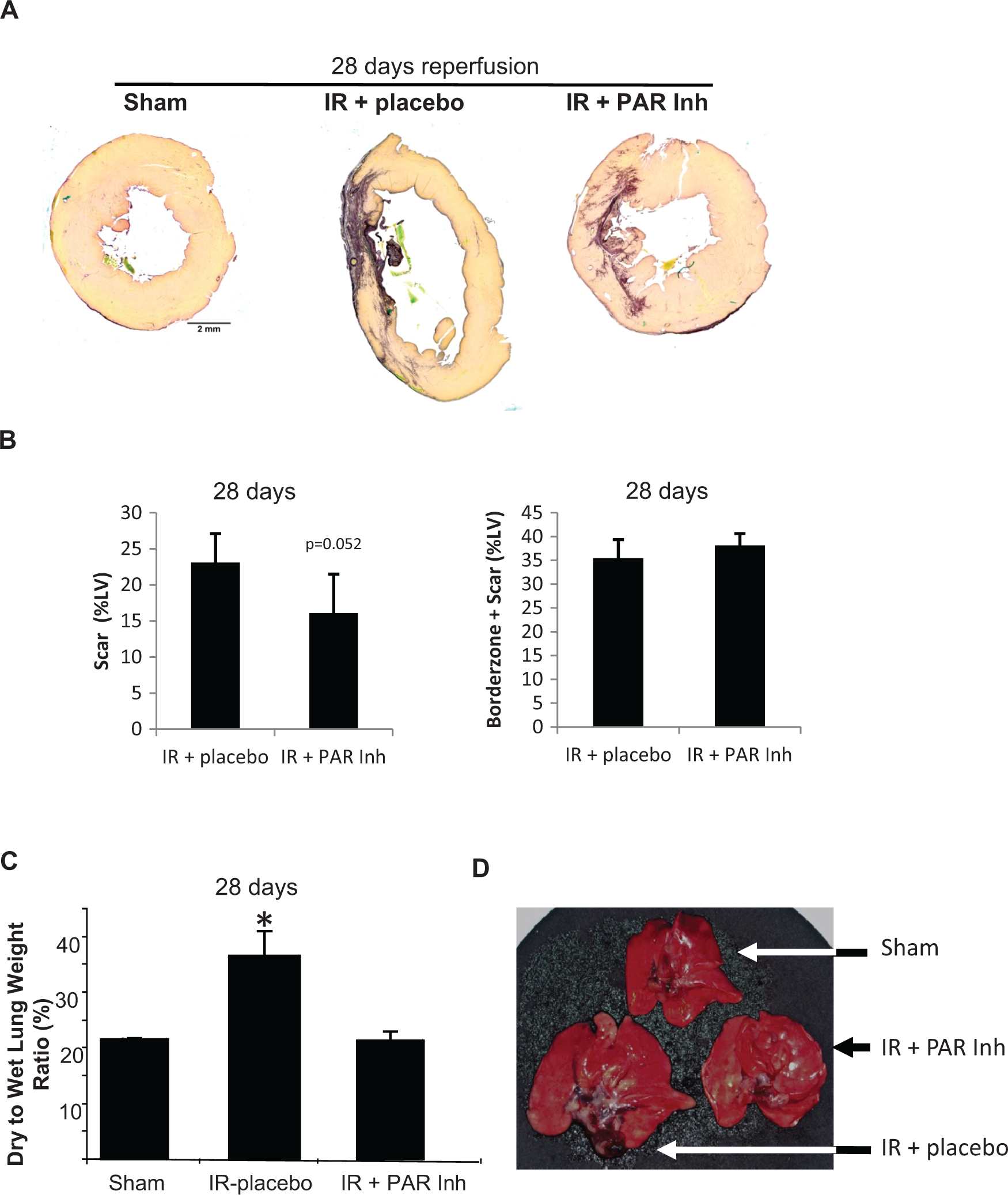
Changes in cardiac fibrosis and pulmonary edema. Rats were subject to 30-minutes left coronary artery occlusion followed by 28 days of reperfusion. The protease-activated receptor (PAR) 1 inhibitor (Inh) or placebo was administered 15 minutes after the onset of reperfusion and continuously through the 28 days. A, Representative cross-sections are shown from the mid-ventricle of Sham, ischemia-reperfusion (IR) + placebo, and IR + PAR Inh group. Calibration Bar = 2 mm. B, Scar Size as a percentage of the left ventricle. Data are mean ± standard error of the mean (SEM), n = 6/group, *P < .05 versus Sham. C, Wet to dry lung weights ratios after 28 days of treatment in the IR + PAR Inh group (n = 9) when compared to IR + placebo (n = 10) and Sham groups (n = 12) *P < .05 versus Sham. D, Representative lungs from a Sham rat (top) compared to an IR + placebo (left) and IR + PAR Inh (right) rat.
There was no detectable change in ERK1/2 phosphorylation by immunoblot analysis of myocardial border zone samples from Sham, Sham + PAR Inh, IR + PAR Inh, and IR groups at 28 days (data not shown). Myocardial TGF-β tissue levels were not significantly different between the groups (data not shown).
The PAR1 Inhibition Does Not Acutely Decrease Myocardial Injury When Given After Ischemia In Vivo
To determine whether the reduction in scar size was due to a decrease in infarct size and tissue necrosis resulting from the initial IR insult, we determined infarct size after 2 hours and 3 days of reperfusion. Postischemic administration of the PAR1 inhibitor had no effect on infarct size after 2 hours of reperfusion (Figure 5A). Likewise, after 3 days of reperfusion with chronic PAR1 inhibition, there was no difference in infarct size (33% ± 13% vs 29.9% ± 9% control; Figure 5B and C). The PAR1 inhibition did not limit the degree of leukocyte infiltration into the border zone (data not shown) or appearance of myofibroblasts at 3 days (Figure 5D).

Protease-activated receptor (PAR) 1 inhibition does not prevent acute or subacute myocardial injury. A, Acute infarct size after 2 hours of reperfusion in ischemia-reperfusion (IR) + placebo versus IR + PAR inhibitor (Inh) groups. Infarct size defined as the ratio of the area of necrosis to the left ventricular (LV) area and expressed as a percentage. Data are mean ± standard error of the mean (SEM; n = 6/group). B, Infarct size after 3 days of reperfusion in IR + placebo versus IR + PAR Inh groups. Infarct size defined as the ratio of the infarct zone to the entire left ventricle and expressed as a percentage. Data are mean ± SEM (n = 12/group). C, Representative cross-sections are shown from the mid-ventricle of Sham, IR + placebo, and IR + PAR Inh group after 30-minute ischemia and 3-day reperfusion. Calibration Bar = 2 mm. D, Identification of myofibroblast in the border zone of sham, IR + placebo, and IR + PAR Inh rat hearts using a fluorescent antibody against α-smooth muscle actin. Calibration Bar = 50 µm.
The PAR1 Activation Promotes a Profibrotic Activity in Adult Cardiac Fibroblasts In Vitro
In freshly isolated adult cardiac fibroblasts, small amounts of α-SMA was detected (Figure 6A). After 24 hours of thrombin (5 U/mL) treatment, the cells express large amounts of highly organized α-SMA (Figure 6B), indicating myofibroblast transformation. The PAR1 inhibition reduced thrombin-induced α-SMA expression (Figure 6C). The addition of the PAR1 inhibitor alone did not affect cellular morphology or α-SMA (data not shown).
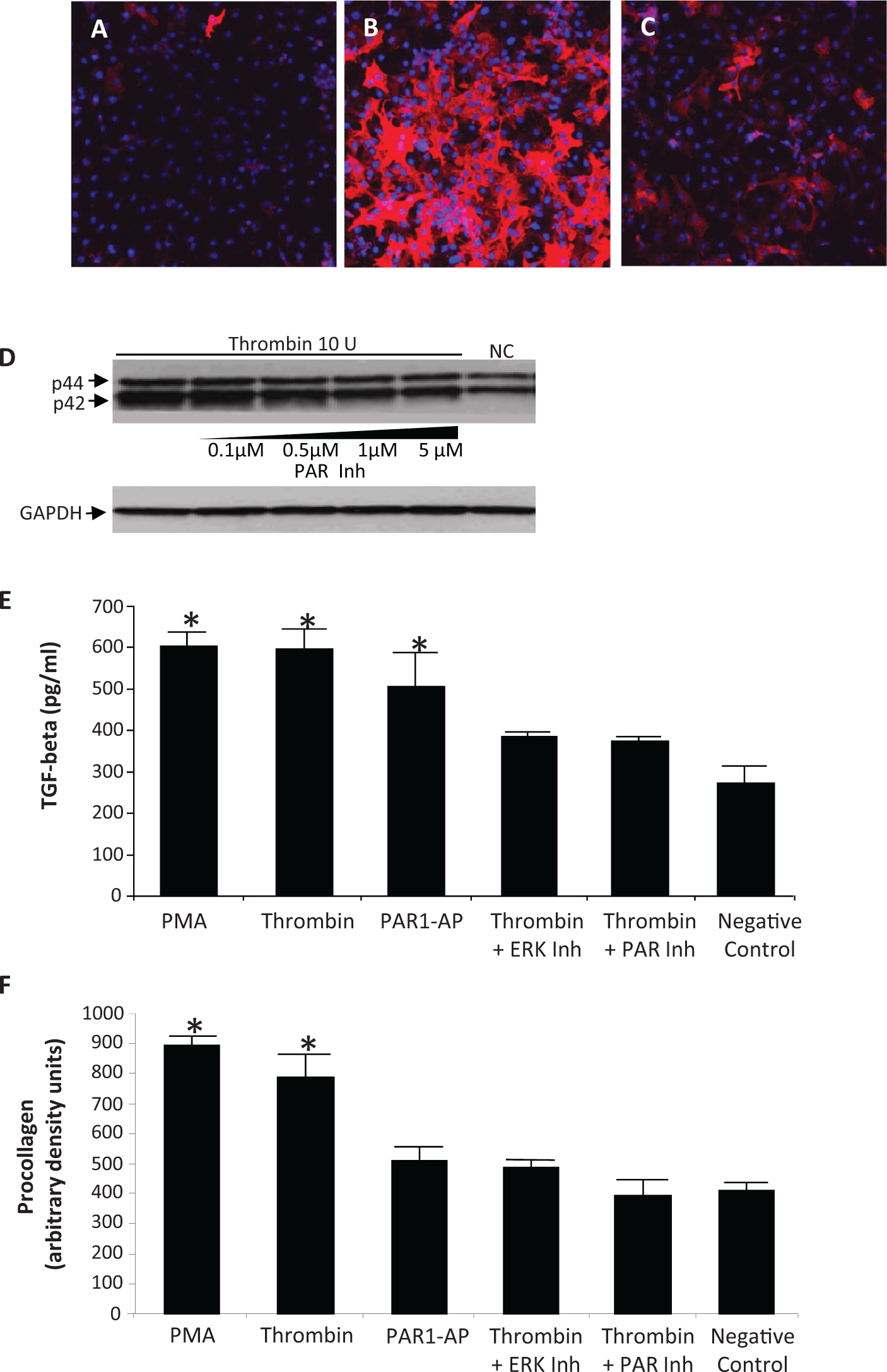
Thrombin stimulation of rat cardiac fibroblasts. Confocal micrographs of freshly isolated adult cardiac fibroblasts treated with phosphate-buffered saline (PBS; A), 5 U/mL thrombin (B), and 5 U/mL thrombin with 1 μmol/L protease-activated receptor (PAR) inhibitor (C) for 24 hours. α-smooth muscle actin was detected using Cy3-labeled antibody (red) and nucleus were stained with DAPI (blue). Thrombin-induced α-smooth muscle actin expression was blocked by the PAR Inhibitor. (D), Western blot analysis showing that thrombin stimulates extracellular signal-regulated kinase (ERK) 1/2 phosphorylation which is diminished by the PAR inhibitor. (E), Enzyme-linked immunosorbent assay (ELISA) analysis showing that thrombin and a selective PAR1 activator peptide (AP) increases transforming growth factor β (TGF-β) secretion. This is blocked by an ERK1/2 inhibitor (PD98059) and a PAR1 inhibitor (SCH79797). Phorbol 12-Myristate 13-Acetate (PMA) was used as a positive control and vehicle was used as the negative control. (F), ELISA analysis showing that thrombin increased procollagen secretion which is blocked by an ERK1/2 and PAR1 inhibitor. Representative blots shown. Data are mean ± standard deviation (n = 3/group). *P < .05 versus negative control.
Thrombin treatment increased the phosphorylation of ERK1/2 after 5 minutes in cultured cardiac fibroblasts which was blocked by increasing concentrations of the PAR1 inhibitor (Figure 6D). After 24 hours, both thrombin and a specific PAR1-activating peptide significantly increased TGF-β secretion, which was blocked by ERK1/2 or PAR1 inhibition (Figure 6E). After 24 hours, thrombin promoted type I procollagen production, which was blocked by ERK1/2 or PAR1 inhibition (Figure 6F). Additionally, the 24-hour thrombin stimulation increased MMP-2 and MMP-9 activity, and this increase was blocked by PAR1 or ERK1/2 inhibition (Figure 7).
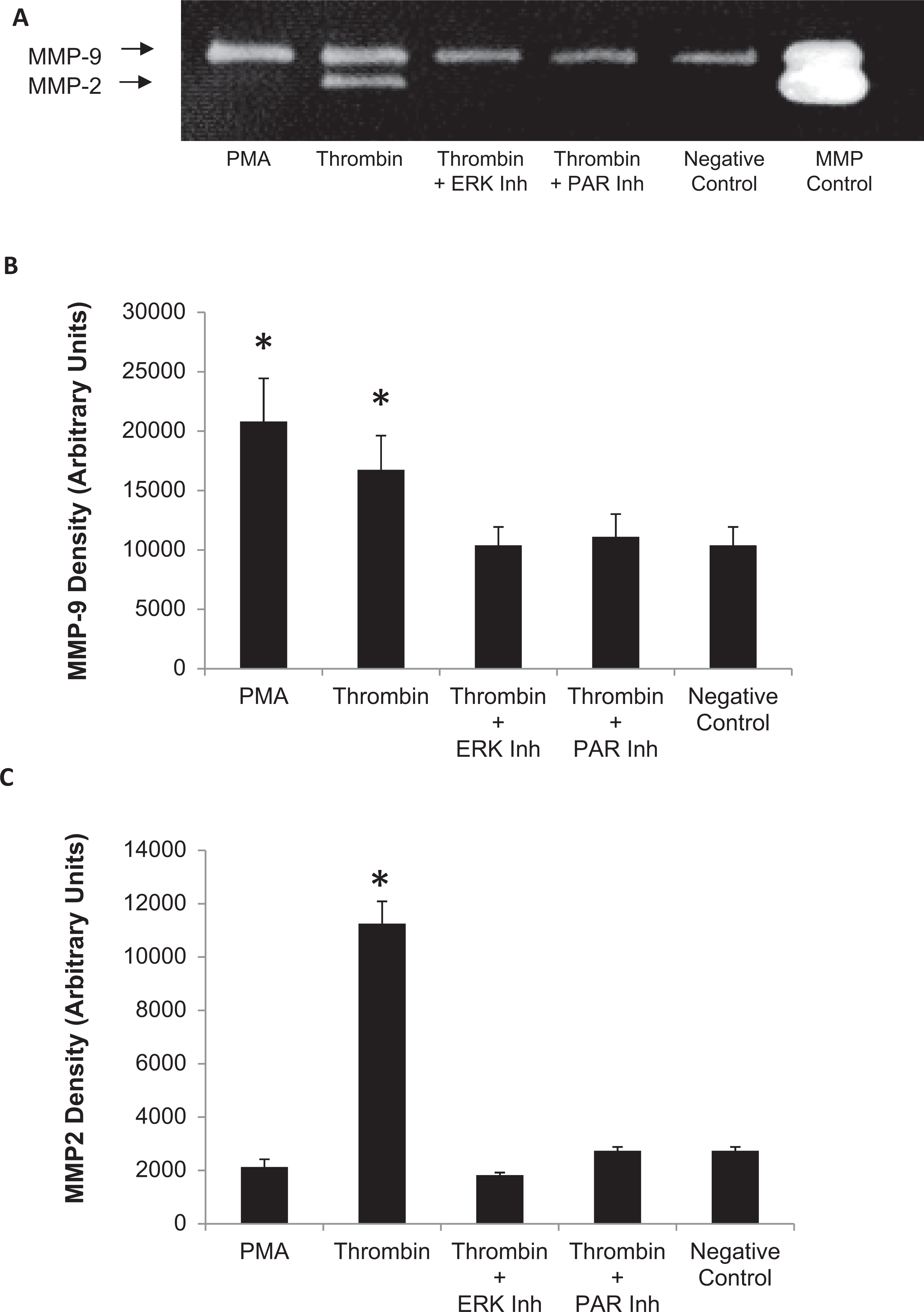
Matrix metalloproteinase (MMP) activity. A, Zymogram analysis showing thrombin activates MMP-2 and MMP-9. This activation is blocked by inhibitors of extracellular signal-regulated kinase (ERK1/2; PD98059) and protease-activated receptor 1 (PAR1; SCH79797). Quantitation of zymolytic bands of MMP-9 (B) and MMP-2 (C). Representative zymogram shown. Data are mean ± standard deviation (n = 3/group), *P < .05 versus negative control.
Effects of PAR1 Activation on 3D Cardiac Fibroblast Tissues In Vitro
In 3D engineered tissues, 24-hour thrombin (10 U/mL) treatment increased cellular contractility and tissue stiffness, indicating the significant remodeling of collagen matrix by cellular activities. 28 The PAR1-activating peptide increased cellular contractility and tissue stiffness, yet the increase was not statistically significant (Figure 1D and E).
Discussion
The major novel finding of this study was that chronic PAR1 inhibition improved LV remodeling and preserved LV function in the reperfused myocardium. After myocardial IR and infiltration of the infarct border zone by neutrophils and macrophages, cardiac fibroblasts transform into myofibroblasts and migrate into the border zone where they promote contraction of the scar. Myofibroblasts are the main source of extracellular matrix proteins in the infarcted and remodeled heart. Selective activation of the TGF-β pathway in the infarct border zone is critically involved in the pathogenesis of fibrotic cardiac remodeling and contributes to the development of cardiac dysfunction following reperfused infarction by enhancing collagen deposition. 29 We now report that several essential phenotypic and functional alterations of cardiac fibroblasts—key players in developing fibrotic remodeling—are dependent on PAR1 activation. In isolated cardiac fibroblasts, thrombin promotes myofibroblast transformation, PAR1- and ERK1/2-dependent upregulation of TGF-β, and procollagen secretion as well as MMP-2 and MMP-9 activation. Furthermore, PAR1 inhibition abolishes the thrombin-mediated contraction and stiffening of 3D engineered cardiac fibroblasts tissues, indicating the functional antagonism of profibrotic events by PAR1 inhibition. These findings implicate that thrombin through PAR1 deregulate matrix synthetic function. The inhibition of PAR1 in vivo results in the reduction of LV replacement fibrosis and remodeling after IR.
Effect of PAR1 Inhibition on Infarct Size and LV remodeling
We have previously shown that acute PAR1 inhibition when given prior to 30-minute ischemia decreases infarct size by 31% after 2 hours of reperfusion. 13 On the contrary, PAR1-deficient mice did not show a reduction in infarct size after IR. 14 These differences may be explained by differences in species or model used or may reflect the ability of the PAR1-deficient mouse to compensate for lack of PAR1 signaling. Regardless, the PAR1 inhibitor when given after the onset of reperfusion did not result in an acute (2 hours) decrease in the infarct size nor did it affect the size of MI and severity of inflammation 3 days later in this study. In addition, our postsurgery mortality was equal between Sham and IR + PAR Inh groups, indicating that even with PAR1 inhibition initial scar formation was adequate to allow wound healing sufficiently to prevent cardiac rupture. Previous studies that used peptide agonists and or PAR1-deficient mice have shown that the effect of thrombin-mediated inflammation is dependent upon PAR1. 30
Sun Y et al observed the appearance of TGF-β mRNA in infarcted rat myocardium on the second day and myofibroblasts on the third day from the beginning of reperfusion. 21 The blockade of PAR1 receptors in our study did not slow appearance of myofibroblasts. It suggests that secretion of TGF-β was sufficient to stimulate the transformation of fibroblasts into myofibroblasts, despite the blockade of PAR1 receptors. This result is consistent with previous studies on transgenic mice with a deficiency of PAR1 receptors in which PAR1−/− fibroblasts did not respond to thrombin but responded to TGF-β. 30
Chronic PAR1 inhibition prevented the dilation of left ventricle. This indicates the important role of PAR1 in the pathogenesis of remodeling in the later stages of healing during myocardial reperfusion. Previous studies have demonstrated that the transition from the phase of inflammation into the proliferative phase manifested with decline in the secretion of TGF-β after 7 days coupled with a decrease in macrophage infiltration 2 weeks after reperfusion. Collagen accumulation at the site begins by day 7. 21 Based on the results of our 28-day observation, the PAR1 inhibitor, although not significantly reducing total infarct size, limited the transmurality of the scar. We speculate that PAR1 inhibition may mediate a reduction in inflammation and collagen deposition after 3 days to attenuate adverse remodeling. However, we cannot disregard the significant increase in mean BP in the IR group when compared to the IR + PAR Inh group. Theoretically, the increased afterload in the placebo-treated animals could also contribute to LV remodeling.
The information obtained by echocardiography substantiates these findings. The LV infarct and associated wall motion abnormalities was confirmed by echocardiography at 3 days. This was associated with a similar decrease in LV EF in the IR and IR + PAR Inh. Early in the course of reperfusion (3 days), PAR1 inhibition was not able to prevent the initial decrease in LV function, but when continued over 28 days, it improved the LV function significantly. The improvement in LV remodeling and function did not result from a reduction in initial infarct size but more likely from a decrease in infarct expansion. The clinical importance of this study lies with the efficacy of postischemic PAR1 inhibition on preserving LV function. Therefore, the PAR1 inhibition may be an effective treatment to improve prognosis after ischemic episodes.
PAR1 Inhibition Decreases a Thrombin-Mediated Fibrotic Phenotype in Cardiac Fibroblasts
A recent study shows that PAR1 is the most abundant G-coupled protein receptor on the cardiac fibroblast, and its activation regulates the myofibroblast transformation. 31 We have shown that PAR1 inhibition attenuated thrombin-stimulated ERK1/2 phosphorylation and TGF-β secretion in cardiac fibroblasts as well as type I procollagen production. The PAR1 inhibition also abolished the thrombin-mediated activation of MMP-2 and MMP-9, the primary MMP involved in extracellular matrix remodeling. The data indicate the importance of ERK1/2 signaling in PAR1-mediated upregulation in tissue-remodeling activities including collagen synthesis and MMP-2/9 enzyme activates by the cardiac fibroblasts. Although the tissue-remodeling activities can be detected biochemically at a cellular level, the detection of their functional phenotypes is not possible by a conventional 2D cell culture system. To determine the functional significance of PAR1-mediated signaling in the tissue remodeling, we used a 3D cell culture system below.
Thrombin stimulation of the 3D engineered tissue increased tissue stiffness and cell contraction, indicating significant remodeling of collagen matrix by cellular activities. These PAR1-mediated tissue remodeling activities suggest that PAR1 signaling is a critical initiator in stiffening the engineered tissues, which conceptually mimic the myocardial stiffening that slows diastolic relaxation. Normalization of the tissue stiffening by the PAR1 inhibitor suggests an inhibition of PAR1 signaling in cardiac fibroblasts may alleviate the tissue stiffening in cardiac fibrosis. Treatment with the PAR1-activating peptide also increases tissue stiffness, although it did not reach significance. This is most likely due to several limitations of the PAR1-activating peptides. Although they are specific for activation of PAR1, they signal preferentially through Gq proteins, whereas thrombin activation of PAR1 signals preferentially through G12/13 proteins and ρ activation. 32,33 In addition, the peptides may not diffuse as readily throughout the 3D tissue culture as the small molecule PAR1 inhibitor.
Although the detectable signaling events in the myocardium at 28 days were unchanged on average, fibroblast-specific signaling could initiate the myocardial remodeling after IR. In our cellular and tissue models, we have shown that thrombin activated PAR1 and its downstream profibrotic activities. However, a detailed mechanism by which PAR1 is activated in vivo after ischemia injury and during remodeling is not clear. Transgenic mice overexpressing PAR1 on cardiomyocytes develop an eccentric cardiac hypertrophy. 14 The deletion of the tissue factor gene in these mice decreases the hypertrophy. This would suggest that low levels of thrombin are generated in the normal myocardium in a tissue factor-dependent manner. 14 The same group showed extravascular fibrin deposition in the rabbit myocardium subject to IR, which suggested coagulation factors gain access to the extravascular space from the damaged endothelial barrier. 34 In addition, MMP-1 has been shown to cleave and activate PAR1. 35 As MMP-1 becomes upregulated in the early stages of repair after ischemia, 21 it may activate PAR1 and contribute to replacement fibrosis and infarct expansion. Nevertheless, the continued activation of PAR1 in the myocardium may be contributing to excess fibrosis and scarring after ischemic injury. Notwithstanding the activation mechanisms, pharmacological inhibition of PAR1 activation or its downstream signaling could be an effective treatment option.
Limitations in Current Analysis
There are several limitations to this study. It is not clear whether in vivo cardioprotection by SCH79797 is shared by other PAR1 antagonists. It has been suggested that SCH79797 may have nonspecific effects. 36 The SCH79797 was characterized initially on human platelets as a competitive inhibitor of the PAR1-selective peptide agonist for binding to the receptor, with IC50 values of 70 nmol/L. 27 De Serio et al found that SCH79797 inhibited the growth of PAR1-null fibroblasts at concentrations exceeding 100 nmol/L. 36 They also found 150 nmol/L SCH79797 inhibited ERK1/2 phosphorylation in PAR1-null fibroblasts stimulated with 10% FBS. 36 In our studies, we used an in vitro and an equivalent in vivo dose of 1 µmol/L, therefore, we cannot rule out nonselective effects of SCH79797. We have previously shown that SCH79797 is a competitive antagonist with a specific PAR1 activating peptide in our IR model, indicating that the effects of SCH79797 are at least in part through PAR1 in this model. 13 Furthermore, in our fibroblast cell culture studies, we have shown SCH79797 is working through PAR1.
Due to limitations in our study protocols, we used different methods to measure infarct size acutely and at 3 and 28 days. We did not design the study to measure AAR at 3 and 28 days, and we used the border zone + scar as a surrogate marker of AAR at 28 days. It is well known that the amount of AAR determines the infarct size. Although our AAR was consistent in our acute studies, and our technique was the same for acute or chronic studies, we cannot exclude the possibility that the AAR was smaller in our chronic studies accounting for the change in infarct size at 28 days. However, since our infarct size did not change at 3 days, most likely the trend toward a smaller scar size at 28 days is due to an inhibition of scar expansion. Furthermore, we were able to verify the infarcted territory by echocardiogram at 3 days. We did not infuse paraffin into the hearts under end-diastolic pressure and because some distortional effects of dehydration and shrinkage associated with embedding could not be avoided, our histological measures may not exactly reflect in vivo changes. Still, we did have a satisfactory correlation between the histological and echocardiography data that we obtained.
We did not assess whether administering the PAR1 inhibitor at a later time point (i.e. 3-7 days after reperfusion) prevents remodeling. Our initiation of treatment occurred within seconds of reperfusion. Another limitation of the present study is the use of otherwise healthy animals to investigate the chronic pathophysiology of IR injury. Most patients with cardiac disease have a variety of risk factors (eg, hypertension, hypercholesterolemia, and diabetes). It may not be possible to realize a significant benefit in the setting of myocardial IR in such individuals. However, since PAR1 expression has been associated with end-stage congestive heart failure, 11,12 PAR1 inhibition may still remain a therapeutic potential.
Conclusions
Previously, PAR1 inhibition has been shown to limit IR injury when rats were treated immediately before and during ischemia injury. 13 Our present data show that the postischemia PAR1 inhibition may not limit initial IR injury but can downregulate the downstream signaling activities of PAR1 in cardiac fibroblasts to attenuate cardiac remodeling and fibrosis. Targeting PAR1 signaling can be a novel strategy to protect against postischemic LV remodeling and failure. Although, there has been no clinical application of the PAR1 inhibitor used in this study (SCH79797) at present; 2 other PAR1 inhibitors, SCH 530348 and E5555, developed for antiplatelet therapy in patients with coronary artery disease are undergoing clinical evaluation. 37,38 We predict these drugs may also prove beneficial in preventing postinfarction cardiomyopathy in patients. Considering that cardiac hypertrophy and failure remains a leading cause of mortality and morbidity worldwide, 39 the need remains great for the identification and development of new therapies for heart disease.
Footnotes
Authors’ Note
This work was done at the Medical College of Wisconsin, Milwaukee, Wisconsin 53226.
Acknowledgments
We thank Dr John Baker for the use of his rat surgical equipment. We also like to thank Daniel Eastwood (Division of Biostatistics, Medical College of Wisconsin) for his biostatics consulting services.
Declaration of Conflicting Interests
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: Tetsuro Wakatsuki is a cofounder of InvivoSciences, LLC. Other authors declared no conflicts of interest.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This research was supported in part by NIH GM087784 grant (to T. Wakatsuki). J. Strande was also supported by the Research and Education Fund, a component of the Advancing a Healthier Wisconsin endowment at the Medical College of Wisconsin. This publication was supported by the National Center for Research Resources and the National Center for Advancing Translational Sciences, National Institutes of Health, through Grant Number UL1RR031973. Its contents are solely the responsibility of the authors and do not necessarily represent the official views of the NIH.