
Abstract
The migration and proliferation of vascular smooth muscle cells (VSMCs) induced by growth factors play a critical role in in-stent stenosis after percutaneous coronary intervention (PCI). The present study tested the hypothesis that sunitinib malate (sunitinib), a tyrosine kinase inhibitor of multiple receptors for growth factors, can reduce neointimal formation after arterial injury in vivo and sought to reveal the underlying mechanism in vitro. Male Wistar rats with balloon-injured carotid arteries were administered either sunitinib or a vehicle orally for 2 weeks. Sunitinib significantly inhibited neointimal hyperplasia relative to control by reducing active cell proliferation. In cultured human aortic smooth muscle cells (HASMCs), sunitinib significantly inhibited platelet-derived growth factor (PDGF)-induced increases of DNA synthesis, cell proliferation, and migration relative to controls as evaluated by [3H] thymidine incorporation, cell number, and the Boyden chamber assay, respectively. Immunoblot analyses showed that sunitinib suppressed phosphorylation of PDGF-BB inducible extracellular signal-regulated kinase and autophosphorylation of PDGF β-receptor, which are the key signaling steps involved in HASMC activation. These results indicate that sunitinib inhibits neointimal formation after arterial injury by suppressing VSMC proliferation and migration presumably through inactivation of PDGF signaling. As such, it may be a potential therapeutic agent, which targets arterial restenosis after PCI.
Introduction
Percutaneous coronary intervention (PCI) has been a standard medical treatment for patients with coronary artery diseases such as acute myocardial infarction or angina pectoris.1,2 The PCI measures include traditional balloon angioplasty, bare-metal stents, and drug-eluting stents (DESs). 3 These procedures have indeed contributed to reduce the mortality of patients with coronary artery disease; however, coronary restenosis is still a major complication of PCI, especially with plain old balloon angioplasty and bare-metal stents. 4 The main pathology of restenosis is the abnormal migration and proliferation of vascular smooth muscle cells (VSMCs), causing intimal thickening and narrowing of the arterial luminal space. 5 In the last decade, to solve this complication, several types of DES have been used successfully to decrease the rate of restenosis. 6 In DES, agents such as sirolimus, paclitaxel, and zotarolimus suppressed proliferation of VSMCs; however, in-stent thrombosis increased as a complication.7–10 Moreover, the prevalence of coronary in-stent restenosis after PCI remains as high as 3% to 20%. 11
Platelet-derived growth factor (PDGF) signaling is an important pathway involved in the pathology of the abnormal VSMC responses as previously described.12,13 Sunitinib malate (sunitinib) is a tyrosine kinase inhibitor of multiple receptors, including PDGF receptors (PDGF-Rs) and vascular endothelial growth factor receptors (VEGF-Rs) that are associated with tumor cell proliferation and angiogenesis. 14 This compound has been used as an anticancer agent against gastrointestinal stromal tumors and metastatic renal cell carcinoma.15,16 However, to our knowledge, sunitinib has not been studied for the treatment of abnormal vascular remodeling after arterial injury in conjunction with any delivery method.
Therefore, the present study investigated the hypothesis that sunitinib may correct abnormal vascular remodeling represented by neointimal thickening after balloon injury and sought to reveal the underlying mechanisms involved in VSMC reactions.
Materials and Methods
Reagents and Antibodies
Sunitinib was provided by Pfizer Inc (New York, New York) or purchased from Carbosynth Ltd (Berkshire, UK). Recombinant growth factors used for in vitro studies were PDGF-AA, PDGF-BB, and heparin-binding Epidermal growth factor (EGF)-like growth factor (HB-EGF; R&D Systems, Minneapolis, Minnesota). Primary antibodies used were proliferating cell nuclear antigen (PCNA; PC10), phospho-p44/42 Mitogen-activated Protein Kinase (MAPK) (Erk1/2; Thr202/Tyr204), phospho-PDGF receptor β (Tyr751), PDGF receptor β, phospho-EGF receptor (Tyr1068), EGF receptor, glyceraldehyde 3-phosphate dehydrogenase (Cell Signaling, Danvers, Massachusetts), and extracellular signal-regulated kinase 1 (ERK1; Santa Cruz Biotechnology, Santa Cruz, California).
Balloon Injury in Rat Carotid Artery and Administration of Sunitinib
Male Wistar rats (Japan Laboratory Animals, Japan, Tokyo) weighing 250 to 300 g were used in this study. Injury of the left common carotid artery was performed as described elsewhere.17,18 Briefly, each rat was anesthetized with an intraperitoneal injection of sodium phentobarbital (64.8 mg/kg), and the left common carotid artery was exposed. A 2F Fogarty balloon catheter (Edwards Lifesciences, Irvine, California) was inserted via the external carotid artery into the common carotid artery and advanced to the aortic arch. The balloon was then inflated with 200 μL of air and pulled back 3 times along the length of the carotid artery after which the balloon catheter was removed and the external carotid artery was ligated. Sunitinib (10 mg/kg per d) or vehicle (20% 2-hyroxypropyl-β-cyclodextrin) was orally administered by gavage to each rat for 14 days until euthanized as per previous rat studies.19,20 All animal protocols were approved by the Institutional Animal Care and Use Committee of Nippon Medical School, Tokyo, Japan.
Histological Analyses of Carotid Artery Cross Sections
At the time the animals were sacrificed, they were perfused through the left ventricle for 5 minutes with 10% neutral phosphate-buffered formalin at a pressure of 110 mm Hg. The damaged left common carotid arteries were excised 2 weeks postinjury, and a specimen was obtained from the middle portion. Paraffin-embedded cross sections (4-µm thick) were stained with hematoxylin-eosin, deparaffinized with xylene, and hydrated in a series of degrading concentrations of ethanol. The arterial sections were then stained with hematoxylin-eosin, elastic Masson trichrome, or immunostained with PCNA antibody for further histological analyses. Three different portions of each artery were selected: proximal, central, and distal portion (2 mm apart each). The intimal and medial areas of each arterial section were assessed with the image-based processing program Image J (National Institute of Health). The mean intimal area, medial area, and intima/media ratio were calculated. The number of PCNA-stained cells was counted in 5 different views. The PCNA-positive ratio (PCNA-positive/total cell nuclei) was also calculated. 21
DNA Synthesis and Number of VSMCs
Human aortic smooth muscle cells (HASMCs) were purchased (LONZA, Basel, Switzerland) and maintained as previously reported. 22 Cells from passage 4 to 5 were used for the experiments. The HASMCs on 48-well plates were stimulated for 18 hours by Dulbecco Modified Eagle Medium (DMEM) containing 1% fetal bovine serum (FBS) with 10 ng/mL of PDGF-AA, PDGF-BB or 25 ng/mL of HB-EGF in the presence or absence of the indicated concentration of sunitinib. For the DNA synthesis assay, [methyl 3 H] thymidine (PerkinElmer Inc, Waltham, Massachusetts) at 1 µCi/mL was added to the media 6 hours before the termination of each experiment. 23 The labeling was stopped by adding 1 mol/L ascorbic acid. The cells were washed twice with ice-cold phosphate-buffered saline (PBS) and twice with ice-cold 10% trichloroacetic acid. Trichloroacetic acid-precipitated materials were solubilized with 250 µL of 0.2 N sodium hydroxide and 0.1% sodium dodecyl sulfate, and mixed with 5 mL of Ultima Gold (PerkinElmer Inc). Radioactivity was measured using a liquid scintillation counter (Aloka Co, Tokyo, Japan).
To evaluate the effect of sunitinib on cell proliferation, cells were treated for an additional 4 days (5 days total). Cell number was then determined by the hemocytometer measurement method as described previously. 24
Migration Assay
A migration assay was performed using a Boyden chamber, with 24-well transwell culture inserts and polycarbonate membranes (8-µm pores; Corning Inc, Tewksbury, Massachusetts). 25 The membrane was coated with type-1 collagen (Nitta Gelatin Inc, Osaka, Japan) for 16 hours and washed with DMEM containing 0.1% bovine serum albumin (BSA) before cell suspension. The cells were allowed to settle in DMEM containing 1% FBS for 1 hour before the addition of agents in the lower chamber. Under basal conditions, the lower chambers were filled with 600 µL of DMEM containing 1% FBS, and HASMCs were then allowed to migrate to the underside of the membrane insert at 37°C in 5% CO2 for 5 hours. After cells on the upper surface of the membrane had been mechanically removed with a cotton swab, migrated cells on the lower surface of the membrane were fixed in methanol, stained with Cyto Quick (Muto Pure Chemicals, Tokyo, Japan), and manually counted from 3 different fields (0.5 mm2/field) with a microscope using the Image J software.
Immunoblot Analysis
Human aortic smooth muscle cells were treated with PDGF-BB or PDGF-AA at a final concentration of 10 ng/mL or HB-EGF at 25 ng/mL with or without the indicated concentration of sunitinib for 15 minutes at room temperature. Cells were placed immediately on ice, washed with ice-cold PBS, and added to 1× radioimmunoprecipitation assay (RIPA) buffer (Cell Signaling) with PhosSTOP (Roche, Basel, Switzerland) containing phosphatase inhibitors. 22
After centrifugation, the supernatants of cell lysates were collected and total protein concentration was measured with a Bicinchoninic acid (BCA) Protein Assay Kit (Thermo Scientific, Rockford, Illinois). Protein (10 µg) from each sample was separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis and transferred to nitrocellulose membranes. The membranes were incubated for 1 hour at room temperature with blocking solution (Tris-buffered saline [TBS] containing 0.1% Tween 20 and 5% milk), followed by overnight incubation at 4°C with TBS containing 0.1% Tween 20 and 5% BSA, along with primary antibodies listed previously. The membranes were then rinsed 4 times with wash buffer (TBS containing 0.1% Tween 20) and incubated for 1 hour at room temperature with horseradish peroxidase-conjugated anti-mouse or anti-rabbit antibodies. The membranes were then rinsed 4 times and the bands were visualized using an an Enhanced ChemiLuminescence (ECL) prime detection system (GE Healthcare, Buckinghamshire, UK). Bands were captured using an LAS image system (FUJI Film, Tokyo, Japan).
Statistical Analysis
Results are expressed as mean ± standard error of the mean. Comparisons of the 2 groups were made with the Student t test. Differences between the other treatment groups were first examined with a 1-way analysis of variance after the normality of the data was tested. Post hoc comparisons were made with Fisher projected least significant difference. All statistics were performed using StatView software (Abacus Concepts, Inc, Berkeley, California). A value of P < .05 was regarded as a significant difference.
Results
Sunitinib Inhibited Neointimal Hypertrophy by Reducing Intimal Cell Proliferation in Balloon-Injured Rat Carotid Arteries
We initially administered sunitinib or vehicle orally to rats for 2 weeks following carotid artery balloon injury to assess whether sunitinib could suppress neointimal thickening in vivo. There was no difference in food intake during treatment or in body weight and lipid and glucose profiles after treatment between the 2 groups of rats (Table 1). Hematoxylin-eosin or elastic Masson trichrome-stained arterial sections revealed that balloon arterial injury caused marked intimal thickening by 2 weeks (Figure 1A). However, 2 weeks of sunitinib treatment significantly inhibited the thickening compared to vehicle (Figure 1A and B). The intima/media ratio was significantly reduced as well (Figure 1B), while no differences were detected in the medial area between treatments (Figure 1B). These results indicate that sunitinib specifically suppresses neointimal hypertrophy and luminal narrowing of the injured artery without affecting the medial size.

Sunitinib attenuation of neointimal hyperplasia in balloon-injured rat carotid arteries. Sunitinib (10 mg/kg per d) or vehicle (20% 2-hydroxypropyl-beta-cyclodextrin) was administered orally to male Wistar rats for 14 days after balloon catheter arterial injury. A, Arterial sections of balloon-injured rat carotid arteries stained with hematoxylin-eosin, elastic Masson trichrome, and immunostained with PCNA. Arrowheads indicate internal elastic lamina. Bar = 50 µm. B, Measurement of carotid artery intima and media areas and intima/media ratio calculation. All values are mean ± SEM (n = 7); *P < .01, †P < .05 versus vehicle. C, Inhibitory effect of sunitinib on PCNA-positive proliferating cells in the neointima of balloon-injured rat carotid arteries. The percentage of PCNA-positive cells is the number of PCNA-positive/total cell nuclei number × 100%. All values are mean ± SEM (n = 3); †P < .05 versus vehicle. PCNA indicates proliferating cell nuclear antigen; SEM, standard error of the mean.
Characteristics of Rats in the Studya

a Body weight was measured and blood samples were collected after 12 hours fasting on the last day of experiment (14 days after arterial injury). Daily food intake per rat was estimated during treatment. All values are mean ± standard error of the mean (n = 7).
Next, we evaluated the effect of sunitinib on cell proliferation in the neointima by PCNA staining of the arterial sections (Figure 1A). Sunitinib significantly reduced the ratio of PCNA-positive cells in the intima compared to the vehicle (Figure 1C).
Sunitinib Reduces DNA Synthesis and Proliferation in VSMCs
To assess the direct effect of sunitinib on the proliferation of VSMCs, we conducted a 3 H thymidine incorporation assay in vitro with cultured HASMCs. After stimulating cells for 24 hours with growth factors such as PDGF-AA, PDGF-BB, or HB-EGF DNA synthesis of HASMCs increased (Figure 2A). Sunitinib treatment significantly suppressed the increased DNA synthesis stimulated by PDGF-AA and PDGF-BB in a concentration-dependent manner (Figure 2A). On the other hand, sunitinib did not affect the increased DNA synthesis by HB-EGF stimulation (Figure 2A).

Inhibitory effect of sunitinib on the growth factor-inducible proliferation of cultured human aortic smooth muscle cells (HASMCs). A, Effect of sunitinib on DNA synthesis induced by PDGF-AA, PDGF-BB, and HB-EGF in cultured HASMCs. The HASMCs were treated with or without the indicated concentration of sunitinib either in the absence or in the presence of growth factors for 24 hours. DNA synthesis was calculated by measuring the incorporation of [ 3 H] thymidine. All values are mean ± SEM (n = 4); *P < .001, †P < .05 versus control, ‡P < .01, §P <.001 versus growth factor stimulation alone. B, Effect of sunitinib on the number of HASMCs stimulated by PDGF-AA, PDGF-BB, and HB-EGF. The HASMCs were treated with or without the indicated concentration of sunitinib either in the absence or in the presence of growth factors for 5 days. All values are mean ± SEM (n = 4); †P < .05 versus control, §P < .001, and ||P < .05 versus growth factor stimulation alone. PDGF indicates platelet-derived growth factor; HB-EGF, heparin-binding-like endothelial growth factors.
To confirm the suppressive effect of sunitinib, we counted cell numbers in the same set of experiments with extended treatment. A 5-day treatment with sunitinib significantly reduced the number of HASMCs under PDGF-BB or PDGF-AA stimulation in a concentration-dependent manner, while it did not change under HB-EGF stimulation (Figure 2B). No significant apoptotic cells were observed during the incubation period (data not shown).
Sunitinib Inhibits Cell Migration of VSMCs Stimulated by PDGFs
The migration of VSMCs from media to intima is another key response following endothelial injury; therefore, we investigated the effect of sunitinib on the migration of HASMCs in vitro. Figure 3A shows representative images of the Boyden chamber membrane stained by Cyto Quick stimulated by PDGF-BB. The PDGF-AA, PDGF-BB, and HB-EGF significantly increased HASMC migration approximately 1.7-, 1.6-, and 1.9-fold of control with 10, 10, and 25 ng/mL, respectively (Figure 3B). Treatment with sunitinib significantly suppressed these increases in migration by 80% with PDGF-AA and PDGF-BB, but not with HB-EGF (Figure 3B).
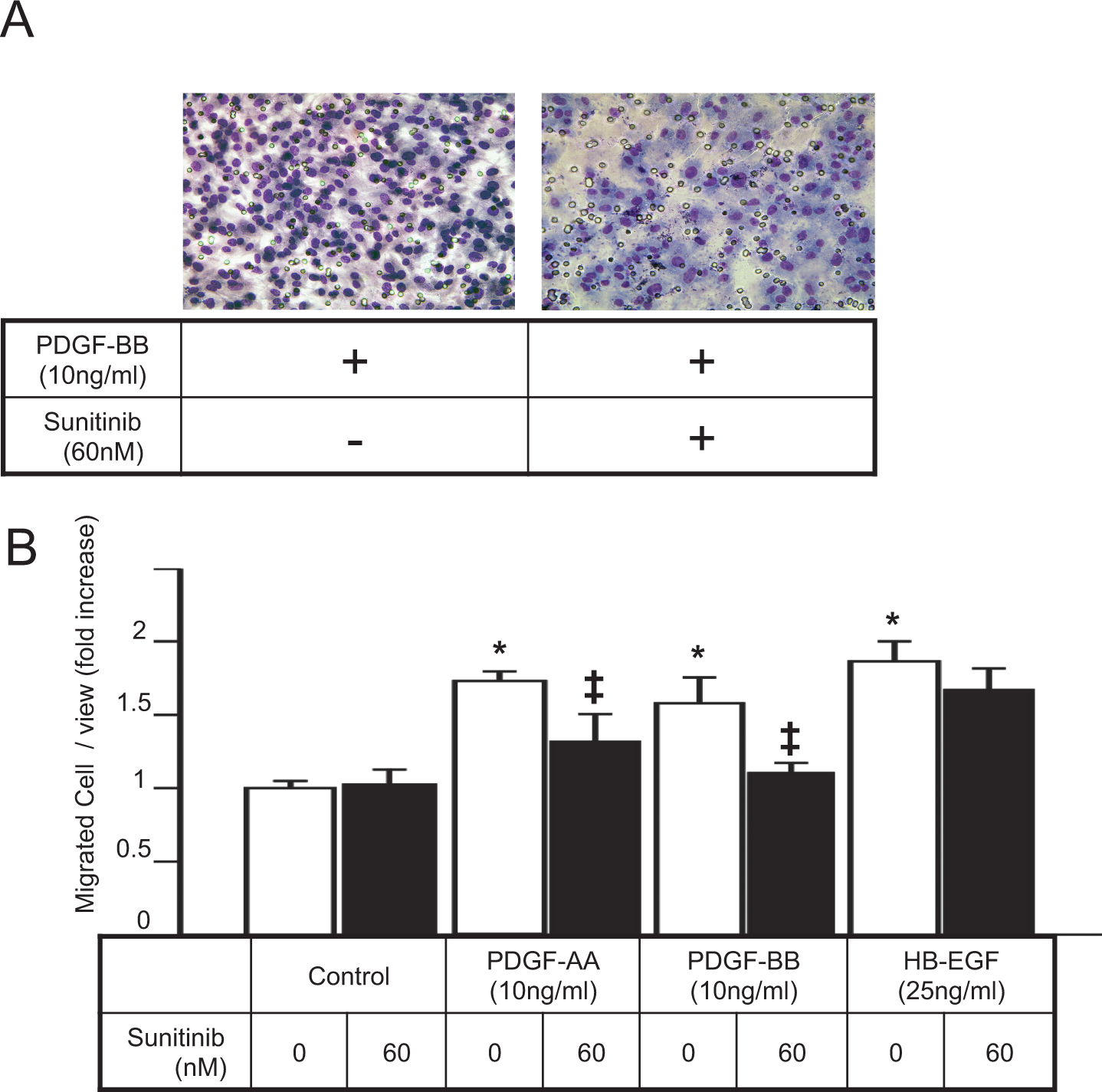
Inhibitory effect of sunitinib on the growth factor-inducible migration of cultured HASMCs. A, Representative image of the Boyden chamber membrane stained by Cyto Quick. The HASMCs were incubated in the Boyden chamber at 37°C for 6 hours with or without sunitinib (60 nmol/L) in the presence of PDGF-BB (10 ng/mL). B, Effect of sunitinib on the migration of cultured HASMCs stimulated by PDGF-AA, PDGF-BB, and HB-EGF. The HASMCs were incubated in the Boyden chamber at 37°C for 6 hours with or without sunitinib (60 nmol/L), in the absence or presence of the indicated concentration of each growth factor. All values are mean ± SEM (n = 4); *P < .001 versus control, ‡P < .05 versus growth factor stimulation alone. PDGF indicates platelet-derived growth factor; HB-EGF, heparin-binding-like endothelial growth factors; HASMCs, human aortic smooth muscle cells.
Sunitinib Suppresses ERK Phosphorylation and Receptor Autophosphorylation in HASMCs Under PDGF Stimulation
We finally examined the effect of sunitinib on growth factor-induced Extracellular Signal-regulated Kinase (ERK) phosphorylation and receptor autophosphorylation by immunoblot analyses to determine the inhibitory mechanism responsible for mitogenic responses in HASMCs. The PDGF-AA, PDGF-BB, and HB-EGF stimulation with concentrations of 10, 10, and 25 ng/mL, respectively, induced ERK phosphorylation. Sunitinib treatment (60 nmol/L) suppressed PDGF-induced, but not HB-EGF-induced, ERK phosphorylation (Figure 4, top 2 panels). In addition, sunitinib canceled the autophosphorylation of PDGF-Rβ induced by PDGF-BB, while no induction was detected by PDGF-AA and HB-EGF (Figure 4, 3rd and 4th panel from top). Moreover, sunitinib did not affect the autophosphorylation of EGF-R induced by HB-EGF (Figure 4, 5th and 6th panel from top).

Inhibitory effect of sunitinib on PDGF- and HB-EGF-inducible intracellular signaling in HASMCs. The HASMCs were stimulated by PDGF-AA, PDGF-BB (10 ng/mL each), or HB-EGF (25 ng/mL) in the presence or absence of sunitinib (60 nmol/L) for 15 minutes. Immunoblot analyses show phosphorylation of ERK, PDGF-receptor beta, and EGF-receptor as well as whole protein levels of those proteins in the treated HASMCs. GAPDH is shown as a loading control. ERK indicates extracellular signal-regulated kinase; PDGF, platelet-derived growth factor; HB-EGF, heparin-binding-like endothelial growth factors; HASMCs, human aortic smooth muscle cells; EGF, endothelial growth factor; GADPH, glyceraldehyde 3-phosphate dehydrogenase.
Discussion
Molecular targeted therapy involves new types of medication that blocks the growth of cancer cells by interfering with specific molecules. This type of therapy developed in the last decade has higher tolerability than traditional treatments. 26 Sunitinib, a small-molecule inhibitor of multiple receptor tyrosine kinase with antiangiogenic and antitumor effects, is one such targeted therapy drug. 14 It suppresses growth factor-induced intracellular signaling, an important pathway, which activates VSMCs in arterial remodeling.
In the present study, we demonstrated for the first time that the administration of sunitinib significantly inhibits abnormal neointimal hyperplasia after arterial injury in an animal model. In the neointima, sunitinib significantly suppressed the rate of PCNA-positive cells compared to vehicle, which indicates a reduced active proliferation of cells. Because there was no difference between the 2 treatments with regard to food intake, body weight, and major metabolic parameters, we assume that sunitinib directly interacts with arterial homeostasis to modify abnormal remodeling.
The migration and proliferation of VSMCs are major pathologies in neointimal thickening following arterial injury. 5 Therefore, we conducted in vitro studies to test the direct effect of sunitinib on such properties using cultured HASMCs. A [ 3 H]-thymidine incorporation assay revealed that sunitinib significantly suppressed DNA synthesis increased by PDGFs. This suppressive effect on the proliferation of HASMCs was confirmed by a decreased number of HASMCs using the same treatment. However, using the same assessment, sunitinib did not affect HASMC proliferation induced by HB-EGF, another mitogenic growth factor.27,28 Moreover, the Boyden chamber assay showed that sunitinib also suppressed HASMC migration induced by PDGFs but not by HB-EGF. These results suggest that sunitinib may significantly and specifically inhibit PDGF-induced VSMC proliferation and migration.
Then, we observed the effect of sunitinib on the growth factor-induced signaling pathways in VSMCs. Immunoblot analyses of HASMCs showed that sunitinib treatment suppressed PDGF-induced ERK phosphorylation and PDGF receptor autophosphorylation that are important signaling responses for VSMC proliferation. However, sunitinib did not affect HB-EGF-signal activation, indicating the selective inhibitory effect of sunitinib on the PDGF signaling cascade.
Sunitinib inhibits tyrosine kinase of VEGF-Rs as well as PDGF-Rs. 14 This effect is different from another PDGF-R tyrosin kinase inhibitor, imatinib mesylate (imatinib). 29 Some, but not all, experimental studies have shown that imatinib reduces neointimal formation by suppressing VSMCs reactions within the injured arterial wall.30–32 It is controversial whether the inhibition of VEGF is protective or not on neointimal hyperplasia.33,34 According to our in vivo experiment, sunitinib treatment suppressed the neointimal hyperplasia, implying that the potential inhibition of VEGF signaling by sunitinib at least did not aggravate the neointimal hyperplasia. However, additional analysis on this issue may be required in the future.
In conclusion, the present study indicates that sunitinib attenuates the pathological activation of VSMCs at least through the suppression of PDGF singaling. All of our findings may not directly apply to human vascular beds with stents, which occur as more complicated pathology by polymer and other components of stents. However, sunitinib may have utility in treating coronary restenosis following PCI.
Footnotes
Acknowledgment
We appreciate Dr Masataka Sata’s (Tokushima University, Japan) technical advice on rat arterial injury and sample preparation.
Authors’ Note
This work was carried out and completed at Department of Bioregulation, Nippon Medical School, Kawasaki, Kanagawa 211-8533, Japan.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was partly supported by a grant from the Takeda Science Foundation to Yoshihisa Okamoto.