
Abstract
We reported that the combination of reactive oxygen species (ROS) quenchers Mn(III) tetrakis (4-benzoic acid) porphyrin (MnTBAP), catalase, and glutathione (MCG) given before 2 hours cold ischemia better protected cardiac mitochondria against cold ischemia and warm reperfusion (IR)-induced damage than MnTBAP alone. Here, we hypothesize that high K+ cardioplegia (CP) plus MCG would provide added protection of mitochondrial bioenergetics and cardiac function against IR injury. Using fluorescence spectrophotometry, we monitored redox balance, ie reduced nicotinamide adenine dinucleotide and flavin adenine dinucleotide (NADH/FAD), superoxide (O2 •−), and mitochondrial Ca2+ (m[Ca2+]) in the left ventricular free wall. Guinea pig isolated hearts were perfused with either Krebs Ringer’s (KR) solution, CP, or CP + MCG, before and during 27°C perfusion followed immediately by 2 hours of global ischemia at 27°C. Drugs were washed out with KR at the onset of 2 hours 37°C reperfusion. After 120 minutes warm reperfusion, myocardial infarction was lowest in the CP + MCG group and highest in the KR group. Developed left ventricular pressure recovery was similar in CP and CP + MCG and was better than in the KR group. O2 •−, m[Ca2+], and NADH/FAD were significantly different between the treatment and KR groups. O2 •− was lower in CP + MCG than in the CP group. This study suggests that CP and ROS quenchers act in parallel to improve mitochondrial function and to provide protection against IR injury at 27°C.
Introduction
Hypothermia is commonly used to protect hearts against subsequent ischemia and reperfusion (IR) injury. Hypothermia, however, is a “two-edged” sword because of its intrinsic deleterious effects that become increasingly greater the lower the temperature. For example, hypothermia causes mitochondrial (m) Ca2+ loading, 1 and when this is coupled to slowed mitochondrial enzyme function as evidenced by increased nicotinamide adenine dinucleotide (NADH) and decreased flavin adenine dinucleotide (FAD),1,2 it may lead to impaired mitochondrial electron transport chain (ETC), which allows electron leak and augments reactive oxygen species (ROS) production. 3 With inefficient quenching of ROS by temperature-sensitive quenchers such as superoxide dismutase (SOD) and catalase, this could further aggravate ROS levels during cold perfusion and may reduce the potentially optimal protective effect of hypothermia. Hence, many currently used cold cardioplegic solutions (CP) contain ROS quenchers,4–7 in order to quench ROS that is generated during ischemia8,9 or on warm reperfusion after cold storage.
In a previous study, 2 we showed that quenching superoxide anion (O2 •−), produced during cold perfusion with normal K+ Krebs Ringer’s solution before cold ischemia, with a cocktail of quenchers, that is Mn(III) tetrakis (4-benzoic acid) porphyrin (MnTBAP), a mitochondrial SOD2 mimetic, plus catalase and glutathione (MCG), provided better mitochondrial preservation, better cardiac function, and reduced infarction. Interestingly, we also observed that quenching O2 •− with MnTBAP alone resulted in less mitochondrial preservation, greater mCa2+ loading, more ROS production, and worse cardiac function compared to the control and the combined cocktail MCG. 2 The implication of the prior study was that O2 •− and its downstream products present during cold perfusion are important factors to be considered during cold storage.
However, in that study, 2 ROS scavengers were added to normal K+ cold Krebs Ringer’s solution, which is not the case in clinical settings where hypothermia is usually combined with CP in the quest that it generates better protection against IR injury than does hypothermia or CP alone.4,6,7 Hence, our aim here was not only to investigate whether adding the cocktail of ROS quenchers (MCG) used in our previous study would have an additive protective effect to cold CP, but also to further investigate whether this possible effect of ROS quenchers would be associated with better preservation of mitochondrial function against IR. To our knowledge, this is the first study to investigate the role of mitochondrial bioenergetics in any additive effect of ROS quenchers plus cold CP in intact beating hearts. We implemented our tools that enable us to assess continuously online changes in mitochondrial redox state (NADH, FAD), mitochondrial ROS emission, and mitochondrial [Ca2+] uptake in the beating heart while undergoing IR.
Materials and Methods
Langendorff Heart Preparation
The investigation conformed to the Guide for the Care and Use of Laboratory Animals published by the NIH (Publication No. 85-23, 1996) and approved by the animal studies committee, Medical College of Wisconsin. Our methods have been described previously in detail.3,10–14 Guinea pigs (n = 73) were anesthetized with ketamine (50 mg/kg, intraperitoneally [IP]), and heparin (5000 U, IP) was administered to prevent clotting. Following decapitation and thoracotomy, hearts were removed and perfused at 55 mm Hg via the aortic root with a Krebs-Ringer’s (KR) solution containing (mmol/L): 138 Na+, 4.5 K+, 1.2 Mg2+, 2.5 Ca2+, 134 Cl−, 14.5 HCO3 −, 1.2 H2PO4 −, 11.5 glucose, 2 pyruvate, 16 mannitol, 0.1 probenecid, 0.05 EDTA, and 5 U/L insulin gassed with ∽97% O2 and ∽3% CO2 (pH 7.4 ± 0.01) at 37°C. Probenecid was added to accentuate loading and intracellular retention of fluorescent dyes. 15 Left ventricular pressure (LVP) was measured with a saline-filled latex balloon inserted into the left ventricle (LV). The volume of the balloon was adjusted so that the diastolic pressure was close to 0 mm Hg at the beginning of each experiment so any later changes will reflect cardiac diastolic contracture due to ischemia. Spontaneous heart rate was monitored with bipolar electrodes placed in the right atrial and ventricular free walls. Coronary flow was measured by an ultrasonic flowmeter (Transonic T106X, Ithaca, New York) placed directly into the aortic inflow line. Cardiac O2 consumption (MVO2) was calculated as coronary flow • heart weight–1 • (Pao2 – Pvo2) • 24 µL O2 / mL (37°C) at 760 mm Hg, and cardiac efficiency as developed LVP • heart rate / MVO2, and %O2 extraction as 100 • (Pao2 – Pvo2) / Pao2 (where Pao2 and Pvo2 are arterial and venous Po2, respectively).
Online Assessment of Mitochondrial Bioenergetic Function
All fluorescence signals were detected in photons/second. All FAD and most NADH molecules are confined to mitochondria.16–18 Most of the O2 •− during cardiac IR injury likely originates from mitochondria while only a small amount is derived from nonmitochondrial sources. 3 It is also possible that nonmyocyte sources such as endothelial and vascular cells can contribute to the total O2 •− generated. However, the overall contribution of this source of ROS is minimal, considering that mitochondrial content of endothelial cells ranges between 2% and 5% 19 while it constitutes 22% to 37% of myocyte volume.20,21 These observations indicate that most of the O2 •− generated in hearts subjected to IR injury originates in mitochondria of cardiac myocytes.
NADH and FAD, m[Ca2+], and O2 •− emission were measured continuously through the LV free wall using fluorescence spectra3,10–14 in different subsets of hearts. A trifurcated fiberoptic probe was placed against the LV to excite and record light signals at specific wavelengths using spectrophotofluorometers (SLM Instruments Inc, Urbana, IL; or PTI, London, Ontario).
In a subset of hearts, as described previously,11,12,22 10 µmol/L dihydroethidium (DHE, Molecular Probes, Eugene, Oregon), a fluorescent probe used to detect the O2 •− radical,9,23,24 was loaded for 20 minutes and washed out (Figure 1B); the LV free wall was excited at 540 nm, and light emission was recorded at 590 nm. O2 •− nonenzymatically converts DHE to 2-hydroxyethidium (2-OH-E+) or a labile precursor that appears to be rapidly made and fluoresces at a slightly shorter wavelength than the heme-peroxidase oxidation product ethidium that can intercalate with DNA.23,24 In other hearts, NADH and FAD autofluorescence was assessed at 350 nm excitation and 450 and 390 nm emissions and at 480 nm excitation and 540 nm emission, respectively.1,8,13,18

A detailed description of the implemented protocol for NADH/FAD (A), superoxide (B), and mitochondrial Ca2+ (C).
Alternatively, other hearts were loaded with 6 µmol/L indo 1 AM (Molecular Probes) for 30 minutes to measure Ca2+ transients at an excitation of 350 nm and emissions recorded at 390 and 450 nm. After initially observing cytosolic Ca2+ transients, hearts were perfused for 15 minutes with 100 µM MnCl2 to quench the cytosolic indo 1 signal.25,26 This was followed by a 15 minute washout (Figure 1C). To estimate actual m[Ca2+], NADH autofluorescence was subtracted from the underlying changes in mCa2+ fluorescent signals for each group.2,8,27 pH in the range of 6.2 to 8.0 does not alter the fluorescence signal in our model. 11 This excludes the possibility of lactic acidosis during ischemia from having a significant effect on fluorescence signals.
Protocol
Hearts were randomly divided into 3 experimental groups: control (CON; KR with 4 mmol/L K+), high K+ cardioplegia (CP; KR with 16 mmol/L K+), and CP plus a combination of ROS quenchers (10 µmol/L MnTBAP, A.G. Scientific Inc, San Diego, California) plus 50 U/mL catalase (Sigma-Aldrich, St. Louis, Missouri) plus 500 µmol/L glutathione (Sigma-Aldrich; CP + MCG). Figure 1 illustrates the implemented protocol for each measured mitochondrial variable. Each heart initially underwent a stabilization period (to reach a rhythmic state of contractility) followed by dye loading to measure either m[Ca2+] or O2 •−. Indo 1 AM caused a small decrease in LVP while DHE caused a small increase in coronary flow. However, these changes in cardiac function were completely reversed upon washout of the unbound dye (data not shown). NADH and FAD autofluorescence were assessed simultaneously. All hearts undergoing ischemia were perfused with either KR alone (CON), CP, or CP + MCG, for 10 min at 37°C, followed by an additional 30 minutes perfusion at 27°C. This was followed by 120 minutes of global 27°C ischemia and 120 min reperfusion at 37°C. At the end of each experiment, the ventricles were cut into 4 to 5 transverse sections of approximately 3 mm and incubated in buffered 0.1% 2,3,5-triphenyltetrazolium chloride to distinguish viable tissue (stained) from necrotic tissue (unstained) for estimating infarct size. 28
Statistical Analysis
All data are expressed as means ± SEM. Among-group data were compared by 2-way ANOVA to determine significance (Super ANOVA 1.11 software; Abacus Concepts, Berkeley, California) at specific times. If F values were significant (P < .05), post hoc comparisons of means tests (Student-Newman-Keuls) were used to compare the groups within each subset. Differences among means were considered statistically significant when P < .05 (2-tailed).
Results
Baseline values were not different among treatment groups. Similar to our previous study, 29 values for all measurements did not change throughout the duration of the time in control experiments (data not shown), which indicates stability of the preparation. Figure 2A and B shows timeline changes in systolic minus diastolic LVP (developed LVP) and diastolic LVP (diaLVP), respectively. All treated hearts were arrested (due to the high [K+] in CP) during the 10-minute period of treatment and throughout the following 27°C perfusion. By the end of 60 minutes, reperfusion-developed LVP was higher in CP and CP + MCG than CON, and diaLVP was lower in the CP and CP + MCG groups compared to the CON group. Values at 120 minutes reperfusion were similar to those at 60 minutes. Cardiac contraction, relaxation, and metabolic data were compared for all groups at 60 minutes reperfusion (Table 1). On reperfusion, each index was better improved in the treatment groups when compared to the CON group. Although some indices in Table 1 are different between CP + MCG and CP alone, these were not statistically significant.
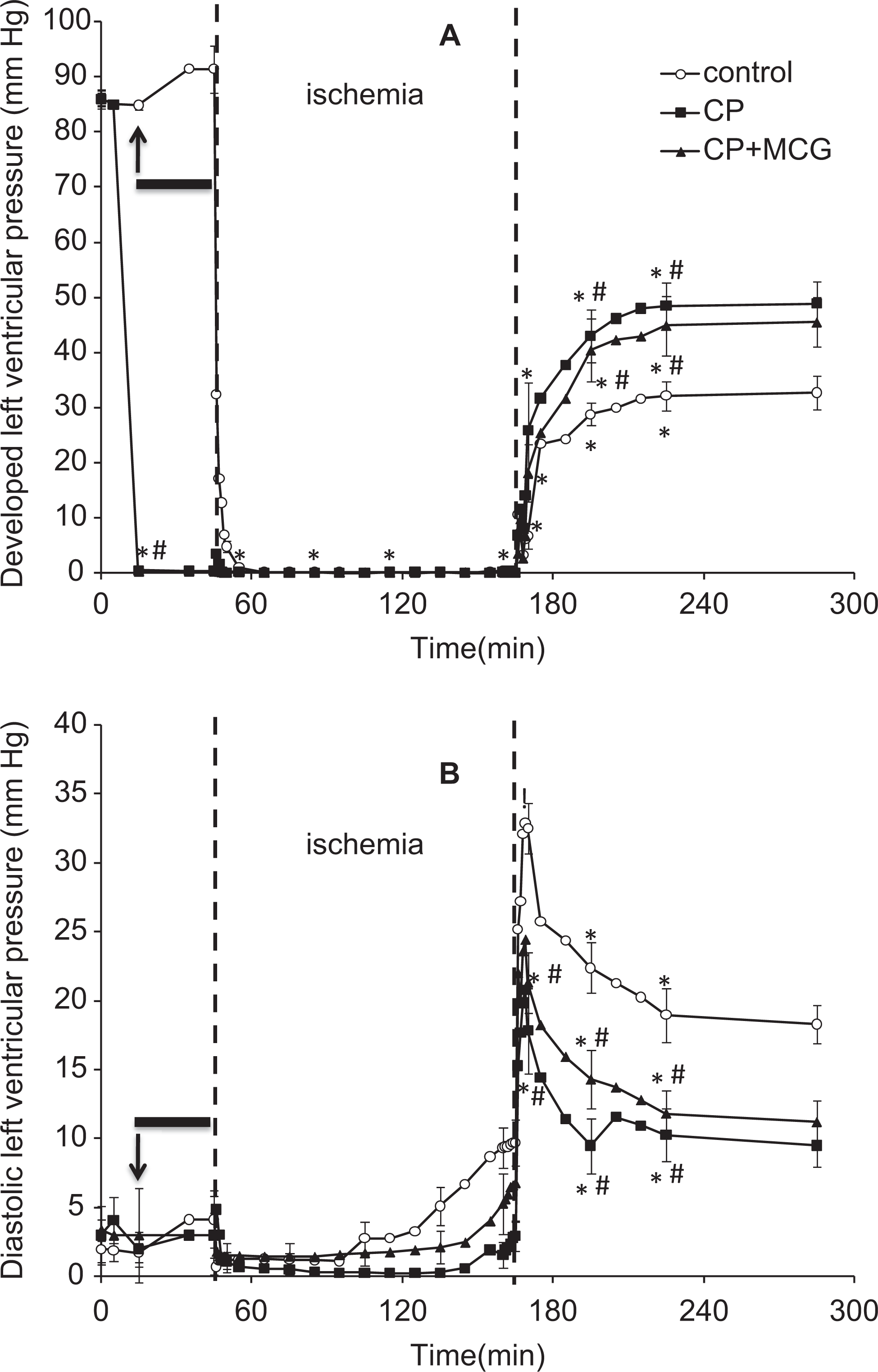
Time course of changes in developed left ventricular pressure (LVP; A) and diastolic LVP (B) at baseline, during treatment, hypothermia, cold ischemia, and warm reperfusion for control (n = 15), cardioplegia (CP; n = 16), and CP + Mn(III) tetrakis (4-benzoic acid) porphyrin, catalase, and glutathione (MCG; n = 14) groups. The arrow indicates where the data point was recorded during treatment at 37°C. The solid horizontal bar indicates the period of treatment at 27°C. The period between the 2 broken vertical lines refers to ischemia. *P < .05 values versus baseline within each group; #P < .05 each treatment versus the control group.
Changes in Heart Rate, Maximal Rates of Contractility (dP/dt max) and Relaxation (dP/dt min), O2 Consumption, % O2 Extraction, and Cardiac Efficiency at Baseline and 60 Minutes Reperfusion a

NOTE: CP = high K+ cardioplegia; CP + MCG = high K+ cardioplegia with MnTBAP, catalase, and glutathione.
aValues are mean ± SEM.
b P < .05 each treatment versus the control group.
Figure 3A and B, respectively, show timeline changes in NADH and FAD. An increase in NADH and a decrease in FAD indicate a more reduced mitochondrial redox state.8,27 NADH increased during treatment with CP and CP + MCG before hypothermia and this was followed by an additional increase during the first 5 minutes of ischemia. Conversely, FAD decreased during treatment with CP and more so during ischemia with CP + MCG. On reperfusion, mitochondrial NADH and FAD were more preserved in the CP and CP + MCG groups compared to the CON group.
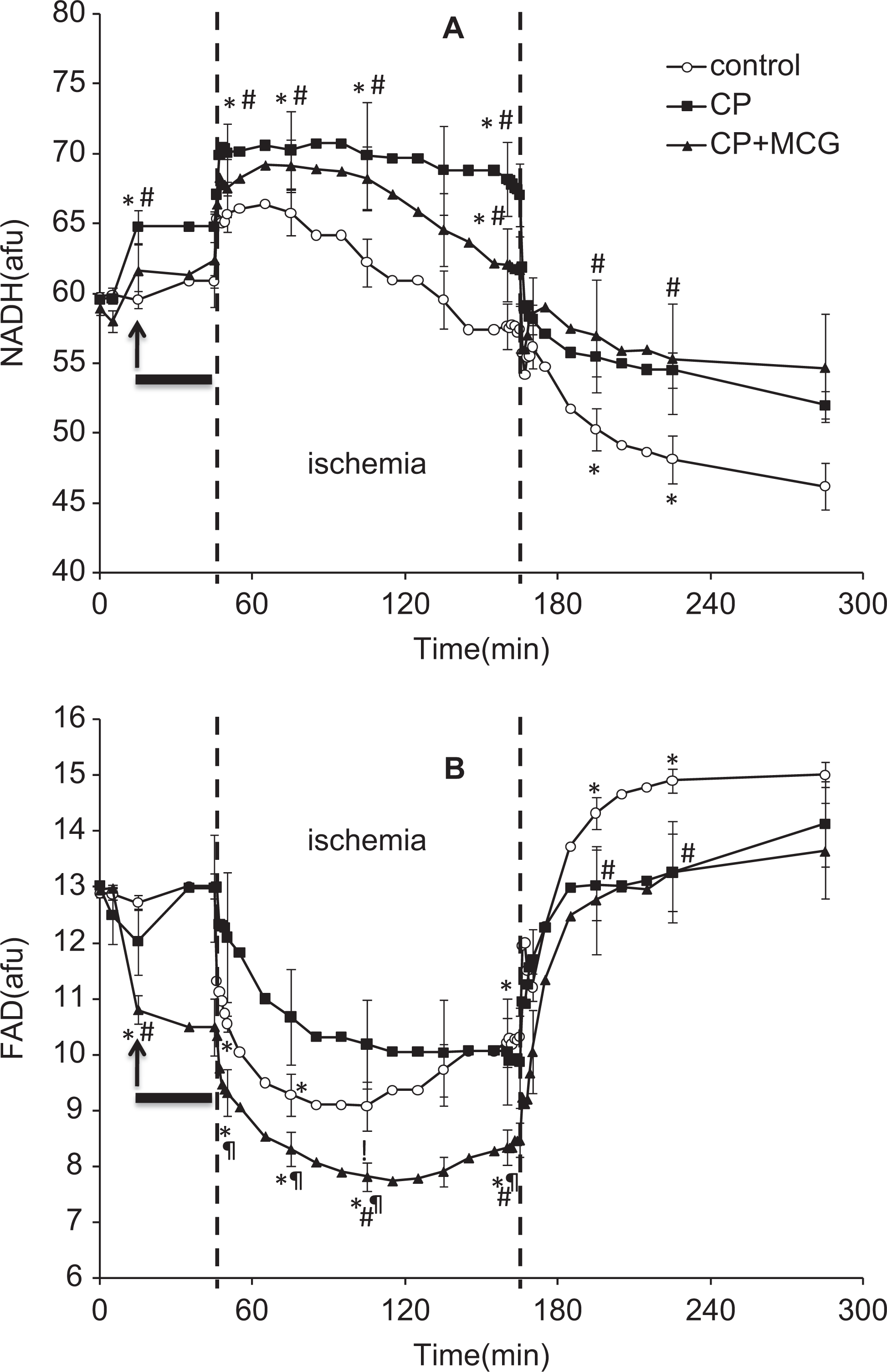
Time course of changes in NADH autofluorescence (A) and FAD autofluorescence (B) at baseline, during treatment, hypothermia, cold ischemia, and warm reperfusion in control (n = 5), cardioplegia (CP; n = 5), and CP + Mn(III) tetrakis (4-benzoic acid) porphyrin, catalase, and glutathione (MCG; n = 5) groups. The arrow indicates where the data point was recorded during treatment at 37°C. The solid horizontal bar indicates the period of treatment at 27°C. The period between the 2 broken vertical lines refers to ischemia. *P < .05 values versus baseline within each group; #P < .05 each treatment versus the control group; ¶P < .05, CP + MCG versus CP alone.
Figure 4A shows timeline changes in O2•− emission in the CON and treatment groups. CP perfusion alone caused a decrease in O2•− emission and cold perfusion increased the O2•− emission slightly in the CON group. During initial ischemia, O2•− emission increased in all groups and surged during the last 20 minutes of ischemia to reach a significantly higher value in the CON group compared to the treatment groups, with CP + MCG having the lowest O2•− emission throughout IR. On reperfusion, the O2•− signal declined in all groups and by the end of reperfusion, O2•− emission was more elevated in CON compared to CP or CP + MCG. Figure 4B shows m[Ca2+] at baseline and at 60 minutes reperfusion. m[Ca2+] was not different among groups during ischemia (data not shown). At 60 minutes reperfusion, CP and CP + MCG treatments resulted in less mCa2+ loading compared to CON, with no significant difference between CP and CP + MCG groups.

A, Time course of changes in superoxide (O2 •−) signal at baseline, during treatment, hypothermia, cold ischemia, and warm reperfusion in control (n = 4), cardioplegia (CP; n = 4), and CP + Mn(III) tetrakis (4-benzoic acid) porphyrin, catalase, and glutathione (MCG; n = 4) groups. The arrow indicates where the data point was recorded during treatment at 37°C. The solid horizontal bar indicates the period of treatment at 27°C. The period between the 2 broken vertical lines refers to ischemia. B, Mitochondrial Ca2+ levels in control (n = 6), CP (n = 7), and CP + MCG (n = 5) groups at baseline and 60 minutes of reperfusion. *P < .05 values versus baseline within each group; #P < .05 each treatment versus the control group; ¶P < .05, CP + MCG versus CP alone.
Figure 5 shows representative images of heart slices stained with TTC to differentiate the infarcted (white) from the viable tissue (red). The figure also shows that CP and CP + MCG groups have smaller infarct sizes compared to CON; CP + MCG showed the smallest infarction but this was not significantly different from CP.

Infarct size as a percentage of total ventricular weight measured after 120 minutes reperfusion from random hearts in control (n = 10), CP (n = 8), and cardioplegia (CP) + Mn(III) tetrakis (4-benzoic acid) porphyrin, catalase, and glutathione (MCG; n = 6). #P < .05 each treatment versus the control group.
Discussion
The major findings of this study are that exposure to cold (27°C) CP before and during ischemia protects hearts in part by preserving mitochondrial redox state (NADH/FAD), lowering m[Ca2+] during reperfusion, and reducing O2 •− emission during IR more than does hypothermia alone. Addition of a combination of ROS quenchers to CP did not further enhance protection against myocardial dysfunction during IR but provided limited improvement against infarction. Mitochondrial dysfunction was reduced with CP treatment with or without ROS quenchers compared to the nontreated, control, hearts. O2 •− emission was lower during IR and FAD remained more reduced during ischemia with CP + MCG compared to CP alone.
Cold CP is widely used for cardiac preservation because it reduces the energy demand that helps to rapidly regenerate ATP on reperfusion. 2 Hypothermia decreases the rate at which intracellular enzymes degrade essential cellular components for organ viability during ischemia. 30 Although hypothermia is the most effective method to preserve hearts during ischemic storage, hypothermia itself has deleterious effects on contractile element and endothelial cell function as the cooling becomes more severe. These defects include cytosolic and mCa2+ loading.1,26 Moreover, hypothermia, per se, without concomitant ischemia increases ROS levels likely by increasing ROS generation and/or reducing ROS scavenging, 3 which may result in mitochondrial and cellular damage proportional to the degree and duration of hypothermia. In a previous study, 2 we showed that the SOD mimetic, MnTBAP, used alone did not protect against ischemic damage due to increased O2 •− downstream products like H2O2 and OH•. But when combined with catalase and glutathione (MCG), cardiac protection was characterized by better normalization of the mitochondrial redox state (NADH and FAD), decreased O2 •− levels and mCa2+ loading during and after ischemia, higher contractile function on reperfusion, and a reduction in infarcted tissue. That study indicated that cardioprotection obtained by mild hypothermia alone during ischemia can be enhanced by selected ROS quenchers administered during the period of cold perfusion before ischemia.
In clinical settings, hypothermic perfusion with ROS scavengers is hardly ever used alone. It is often combined with constantly modified CP solutions in order to achieve the optimal cardiac preservation with good post-ischemic recovery.31–35 Hence, in the current study, we investigated whether addition of ROS quenchers to cold CP before ischemia would have any additive effect on cardiac protection, and more importantly, whether this additive protection is associated with a reduction in mitochondrial ROS and/or improved redox state. Indeed, our data show that the equal cardiac functional recovery in the treatment groups after 60 minutes of reperfusion was associated with similar preservation of mitochondrial redox state (NADH/FAD).
In this study, we used the ROS quencher cocktail MCG because it showed the best protection against cold-induced ROS generation in our previous study. 2 Although adding MCG to CP did not further improve cardiac recovery after 2 hours cold ischemia more than did CP alone, it decreased infarct size. The rationale for using ROS quenchers with cold CP was to reduce ROS levels induced by hypothermia. However, our results show that CP alone reduced O2 •− emission during ischemia compared to control, and at 60 minutes reperfusion, O2 •− emission in the CP group was not different from that in the CP + MCG group. It is unclear how CP alone is effective in decreasing ROS. It is possible that electron flow through the ETC is diminished due to less ATP demand in CP-treated hearts, which could result in less electron leak and lower O2 •− levels.
Another possibility could be simply that there is more effective ROS scavenging by the endogenous glutathione in the CP group. Glutathione is highly regulated by the cytosolic redox state and is rapidly taken up from the cytosol via the decarboxylate and 2-oxoglutarate transporters.36–38 In this way, it effectively links changes in the cellular redox state.37,38 Indeed, one defense against IR-induced ROS accumulation and damage may involve preservation of the NAD(P)H pool. The NADH/NAD+ level through the NADH kinase and transhydrogenase39,40-dependent mechanism maintains the mitochondrial NAD(P)H pool required to maintain the redox status necessary for effective scavenging. Thus, an increase in NADH, as was observed in the CP group, would correlate with increased NAD(P)H-dependent redox scavenging by glutathione.
Lastly, it is unlikely that CP reduced the ROS level indirectly by reducing m[Ca2+] since m[Ca2+] was not different between CP and CON groups during ischemia (data not shown). Although high m[Ca2+] during ischemia is believed to be an instigator of IR injury, it seems that the most damaging Ca2+ influx occurs on reperfusion. 41 All together, it appears that addition of MCG to CP, although it decreased ROS even more than did CP alone, did not markedly enhance CP protection, which may indicate that not all the ROS generated during ischemia is damaging.
Our results showing that addition of MCG to CP did not improve cardiac recovery when compared to CP alone are inconsistent with previous studies4,42–48 that showed improvement upon adding ROS quenchers. It is evident that the production of ROS by cardiac mitochondria in our model during cold ischemia is substantially reduced by CP alone so that the added benefit of ROS quenchers is small. In contrast, it is unlikely that the 2 hours cold ischemia was deleterious enough so that the doses we used were not enough to reduce damage since these drugs showed a modicum of protection in our previous study. 2 Another important consideration is the efficacy of ROS quenchers during ischemia. Reactive oxygen species are generated during ischemia and especially during late ischemia. However, it was reported that superoxide dismutase and catalase may not be as effective during this period since ROS scavenging enzymes are believed to provide the most benefit when O2 is restored to previously ischemic tissue. 49 Although CP + MCG reduced O2 •− emission even more than did CP alone (Figure 4A), it is possible that the surge in ROS during late ischemia, when ROS quenchers are less effective, lessens the beneficial outcome.
In summary, our data show that cold CP alone before 2 hours cold ischemia preserved mitochondrial redox state (NADH, FAD), lowered ROS during subsequent cold IR, and lowered m[Ca2+] during reperfusion. Adding ROS quenchers to the CP provided added protection in reduced infarct size and the better preservation of mitochondrial bioenergetics as shown by the decrease in O2 •− emission during ischemia.
Footnotes
Acknowledgement
The authors wish to thank Anita Tredeau and Steven Contney for their valuable assistance.
The author(s) declared no potential conflicts of interest with respect to the authorship and/or publication of this article.
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was conducted in the Department of Anesthesiology and Anesthesia Research at the Medical College of Wisconsin and was supported in part by the National Institutes of Health (K01 HL73246 to AKSC, R01 HL089514 to DFS); and the Veterans Administration (VA Merit 8204-05P to DFS).