
Abstract
The canonical role of superoxide dismutase 1 (SOD1) is as an antioxidant enzyme protecting the cell from reactive oxygen species toxicity. SOD1 was also the first gene in which mutations were found to be causative for the neurodegenerative disease amyotrophic lateral sclerosis (ALS), more than 20 years ago. ALS is a relentless and incurable mid-life onset disease, which starts with a progressive paralysis and usually leads to death within 3 to 5 years of diagnosis; in the majority of cases, the intellect appears to remain intact while the motor system degenerates. It rapidly became clear that when mutated SOD1 takes on a toxic gain of function in ALS. However, this novel function remains unknown and many cellular systems have been implicated in disease. Now it seems that SOD1 may play a rather larger role in the cell than originally realized, including as a key modulator of glucose signaling (at least so far in yeast) and in RNA binding. Here, we consider some of the new findings for SOD1 in health and disease, which may shed light on how single amino acid changes at sites throughout this protein can cause devastating neurodegeneration in the mammalian motor system.
Introduction
Amyotrophic lateral sclerosis (ALS) is a fatal adult-onset neurodegenerative disease characterized by degeneration and death of upper and lower motor neurons (MNs). Patients typically suffer from progressive motor weakness, which starts focally and spreads through the body leading to paralysis and ultimately death within a few years of diagnosis. Although usually sporadic (without a family history), approximately 10% of ALS cases are familial (fALS) and of those, ~20% are caused by mutations in the gene superoxide dismutase 1 (SOD1) (Kiernan and others 2012). More than 160 mutations in SOD1 have been identified in ALS sufferers, the majority of which are missense point mutations resulting in a dominant mode of inheritance. At least 75 of SOD1’s 154 amino acids have been reported as mutated in ALS and their positions are scattered throughout the five exons of the gene (Saccon and others 2013).
SOD1 is highly conserved throughout evolution (Wang and others 2006), ubiquitously expressed and makes up 1-2% of the total soluble protein in the central nervous system (Pardo and others 1995). Its primary function is thought to be as a cytosolic and mitochondrial antioxidant enzyme, converting superoxide to molecular oxygen and hydrogen peroxide; however, in yeast at least, less than 1% of total SOD1 is required to carry out this canonical function (Corson and others 1998; Reddi and Culotta 2013) leaving the question open as to whether SOD1 plays other equally important role(s), which might account for its abundance.
Many lines of evidence have led to the conclusion that mutations in SOD1 cause ALS via an as yet unidentified gain of function, although it has been proposed that a loss of function may also play a secondary role in disease, at least in some cases (Saccon and others 2013). A great number of cellular mechanisms has been implicated as potentially involved in SOD1-fALS pathogenesis, however, distinguishing cause from effect and identifying the critical processes remains challenging (Redler and Dokholyan 2012). Here, we describe emerging themes in SOD1 biology that suggest this enzyme is involved in a widening array of cellular processes in both health and disease; this may shed new light on the pathogenesis of SOD1-ALS.
Transmission of SOD1
Some causative proteins for neurodegenerative diseases may have “prion-like” properties: the ability to sequester wildtype proteins and seed their aggregation or misfolding, and to act as transmissible agents between cells (Polymenidou and Cleveland 2012) (Fig. 1). SOD1 displays prion-like properties in vitro and in cellular and now in animal models, which is particularly of interest for SOD1-fALS, given the focal start of ALS, the finding that motor neuron death is not cell-autonomous (e.g., see Ilieva and others 2009), and the implications for therapy to halt the spread of pathology. These findings are also important for a potential mechanism for spread, and possibly even pathogenesis in sporadic ALS (sALS), given the presence of aggregated SOD1 in sALS cases (Bosco and others 2010; Forsberg and others 2010; Grad and Cashman 2014).
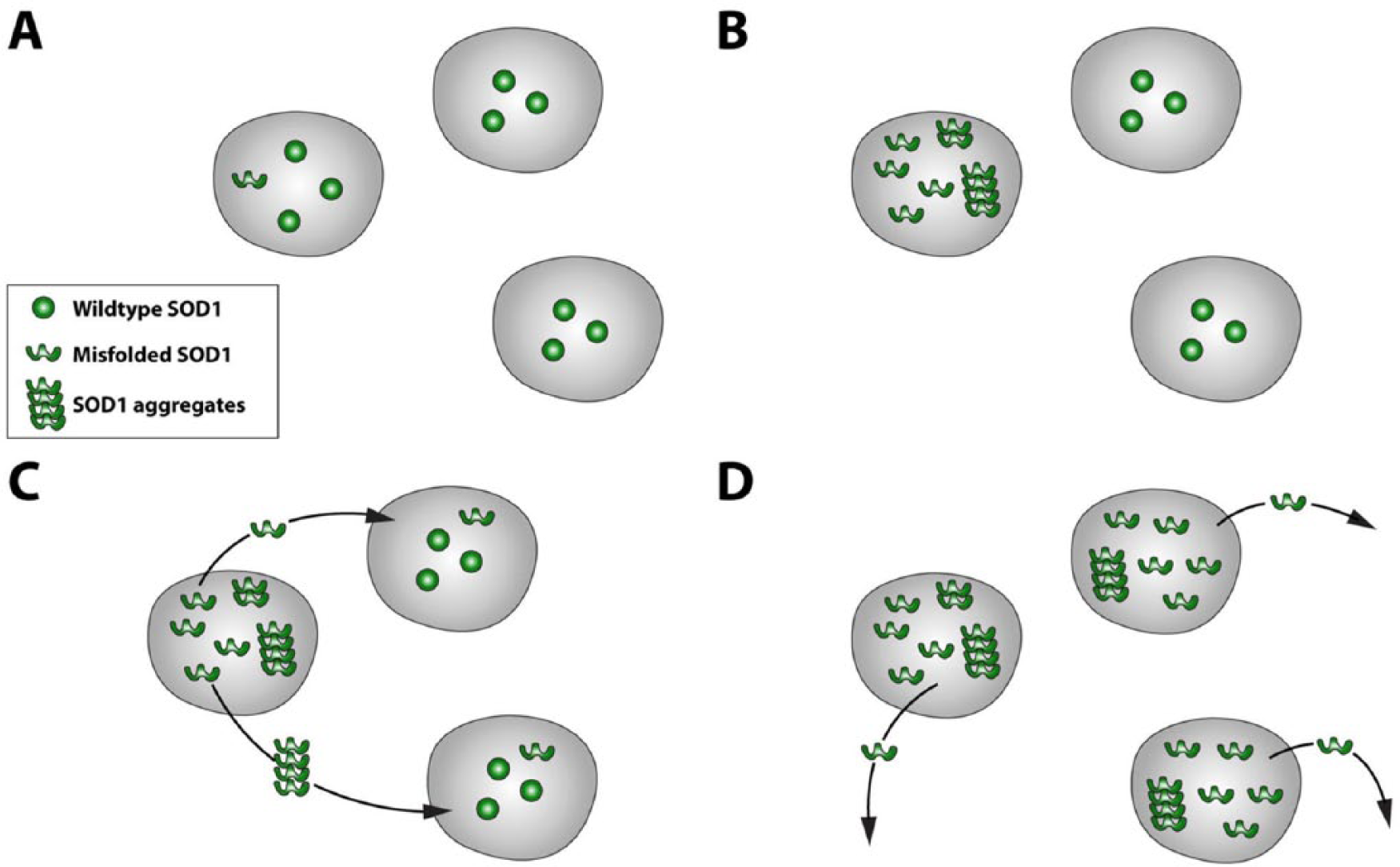
Superoxide dismutase 1 (SOD1) may have prion-like properties. (A) Misfolded SOD1 within a cell could (B) sequester and misfold wildtype SOD1, ultimately producing aggregates, and (C) if secreted and taken up by neighboring cells could (D) cause a “chain reaction” of misfolding, aggregate formation, and transmission in a prion-like manner. The exact nature of this process—for example, the roles of monomers and aggregates—remains unknown.
SOD1 Seeded Aggregation In Vitro
In vitro assays measuring aggregation propensity show recombinant wildtype and ALS-mutant SOD1 can seed aggregation of themselves (self-seeding) and of each other (cross-seeding). Seeded amyloid fibrilization of SOD1 occurred at acidic pH and in the presence of a chaotrope (Chia and others 2010), and non-amyloid seeded fibrilization was seen at physiological pH (Hwang and others 2010). Spinal cord tissue homogenate from a SOD1G93A transgenic mouse (which models human ALS) was then shown to efficiently self- and cross-seed the amyloid fibrilization of recombinant wildtype and mutant SOD1 in vitro (Chia and others 2010). However, self- and cross-seeding does not, alone, indicate prion-like properties; the prion-like nature of a protein also comes from its ability to transmit between cells and indeed between organisms.
Transmission in Cell Models
Mutant SOD1 is found in medium from primary cultures of whole spinal cord and of astrocytes from SOD1-ALS transgenic mice (Basso and others 2013; Urushitani and others 2006). Furthermore, mutant SOD1 is secreted in exosomes from primary astrocytes and neuronal-like stable cell lines, (Basso and others 2013; Grad and Cashman 2014) (Fig. 2). Thus there is a mechanism for mutant SOD1 to get out of cells.
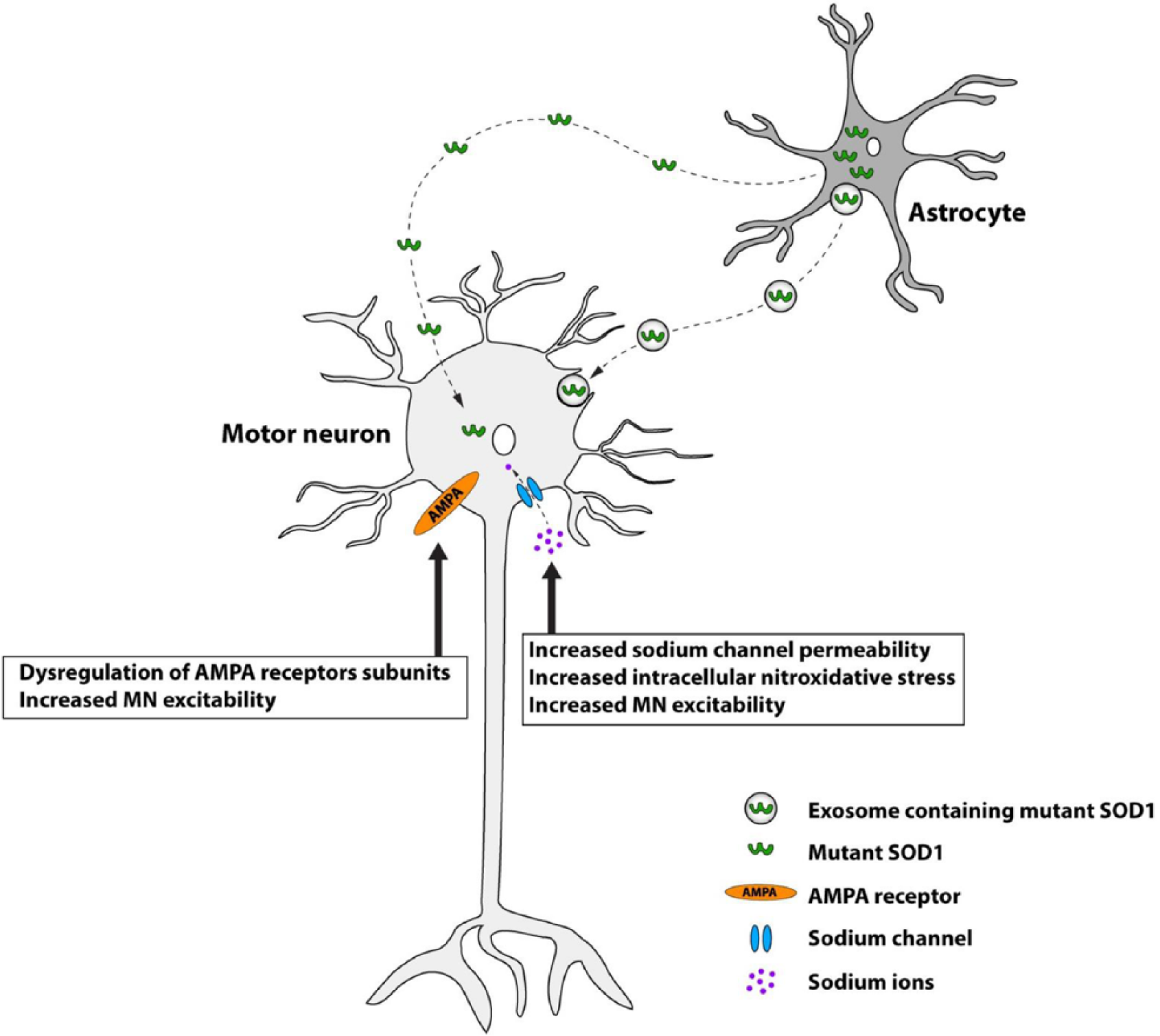
Mutant superoxide dismutase 1 (SOD1) may be transmitted from astrocytes to motor neurons, causing cell death. Mutant SOD1 is secreted in exosomes by astrocytes and may be taken in by motor neurons; propagation of misfolding may then proceed within the cell (see Fig. 1). Exposure of motor neurons to mutant SOD1 results in increased sodium channel permeability, induction of nitroxidative stress, hyperexcitability likely through dysregulation of AMPA receptors as well as sodium channel dysfunction and reduced viability.
In terms of getting into cells, a range of recombinant forms of SOD1 (including wildtype and mutant, aggregated and non-aggregated) are efficiently internalized by neuronal-like stable cell lines, via the non-selective mechanism, macropinocytosis (Grad and Cashman 2014; Münch and others 2011; Sundaramoorthy and others 2013). Primary mouse spinal cord cultures and neuronal-like cell lines also take in wildtype and mutant SOD1 via internalization of exosomes (Basso and others 2013; Grad and others 2014).
Once internalized, recombinant aggregated mutant SOD1 can self-seed aggregation of stably expressed mutant SOD1; this is sustained for up to 1 month and through multiple passages of a mouse neuroblastoma-derived cell line, long after the exogenous seeds had apparently been cleared from the cells (Münch and others 2011). Furthermore, recombinant mutant SOD1 (both aggregated and non-aggregated forms) can also cross-seed endogenous wildtype SOD1—which can also be self-seeded by recombinant aggregated wildtype SOD1 (Sundaramoorthy and others 2013).
Self-seeded aggregation of endogenous SOD1 by conditioned medium has been shown in stable cell lines through multiple passages; this was blocked by both SOD1 knock-down and immuno-depletion of misfolded SOD1 in the medium (Grad and others 2011; Grad and Cashman 2014).
Transmission between Mouse Models
Recently, transmissibility of mutant SOD1 and motor neuron disease pathology between different transgenic mouse models appears to have been demonstrated. Spinal cord homogenates from SOD1G93A mice were injected into the spinal cords of different SOD1 transgenic models and motor neuron disease was induced in an otherwise unaffected transgenic animal expressing low levels of a different (SOD1 G85R) mutant SOD1 protein (Ayers and others 2014). Remarkably, when tissues from the induced mice were used to inoculate new SOD1G85R mice (“second passage homogenates”) disease onset was earlier in recipient animals, exactly as seen in prion disease models. Other data shows potential SOD1 “strains” akin to prion strains (Ayers and others 2014). This article offers compelling data to indicate the physiological prion-like transmission of mutant SOD1 and motor neuron disease pathology in vivo. Of note, one-third of the SOD1G85R mice inoculated with homogenates from wildtype SOD1 transgenic mice also developed the motor neuron degeneration phenotype, while self-seeding experiments in two other mutant SOD1 transgenic lines failed to accelerate disease in these mice. Future work to identify whether a misfolded SOD1 species is both necessary and sufficient to induce disease, and to determine whether the phenomenon is applicable to other SOD1 variants will be needed.
Mutant SOD1 Expressing Glia Are Toxic to Motor Neurons
Data from transgenic mice with tissue specific/restricted expression of mutant SOD1 demonstrate that non-neuronal cells are involved in the progression of disease-related phenotypes (reviewed in Ilieva and others 2009). In vitro experiments also clearly demonstrate the toxicity of mutant SOD1-expressing glia to motor neurons.
Astrocytes
Toxicity of astrocytes, to motor neurons has been demonstrated by co-culture, and by the application of astrocyte conditioned medium (ACM) (Basso and others 2013; Haidet-Phillips and others 2011; Meyer and others 2014; Nagai and others 2007). Intriguingly, this interaction appears to be cell-type specific: mutant SOD1 expressing primary astrocytes reduced viability of both primary and embryonic stem cell-derived MNs in co-culture, but interneurons, GABAergic neurons, or dorsal root ganglion neurons were not affected. Mutant SOD1 fibroblasts, microglia, cortical neurons, or myocytes were not toxic to co-cultured MNs (Nagai and others 2007).
Induction of nitroxidative stress and hyperexcitability has been proposed as a possible mode of astrocyte toxicity to motor neurons: within 30 minutes of applying mutant SOD1 ACM, primary spinal MNs had increased excitability and increased sodium channel permeability; this exposure, followed by 4 days of culture with normal medium, was sufficient to cause ~50% MN death (Fritz and others 2013). ACM also produced an increase in intracellular nitroxidative stress in cultured MNs (Rojas and others 2014) (Fig. 2). Similarly, co-treatment with ACM and sodium channel blockers protected MNs from hyperexcitability, nitroxidative stress, and cell death, while antioxidants protected against nitroxidative stress and significantly improved MN survival, although their effect on sodium channel activity was not assessed (Fritz and others 2013; Rojas and others 2014). MNs co-cultured with mutant SOD1 expressing astrocytes have been shown to have dysregulated AMPA receptor subunits and increased excitability (Van Damme and others 2007).
However, conversely, a separate study of mutant SOD1 transgenic mouse models found MNs with lowest basal excitability were the most vulnerable, and pharmacologically increasing MN activity reduced misfolded SOD1 pathology and markers of cellular stress while decreasing activity had the opposite, detrimental, effects (Saxena and others 2013).
The toxic factor released by astrocytes has not yet been identified and may well be/include mutant SOD1 because exosome depletion prevents ACM toxicity (Basso and others 2013). Interestingly, astrocytes from mutant TDP43 transgenic mice, sALS and C9orf72 expansion patients have also been shown to cause MN toxicity by co-culture and by ACM (Haidet-Phillips and others 2011; Meyer and others 2014; Rojas and others 2014) suggesting some common toxic pathway in sALS and fALS.
Microglia
Activated microglia are thought to play an initially protective role in ALS, but then possibly become toxic due to increasing neuroinflammatory processes as disease advances (Lewis and others 2012). Microglia purified from mutant SOD1 mice are toxic to stem cell–derived human MNs and this toxicity appears to be dependent on prostaglandin signalling: activation of prostaglandin D2 (PGD2) receptor 1 (DP1) in wildtype mouse and human microglia prior to co-culture with MNs, induced significant MN toxicity. Conversely, pretreatment of mutant SOD1 mouse microglia with a pharmacological inactivator of DP1, or genetic ablation of DP1 expression, significantly attenuates toxicity to MNs (de Boer and others 2014).
Expression of mutant SOD1 up-regulates PGD2 receptor 1 (PTGDR) transcript levels in mouse and human microglia, and increases the release of PGD2 in rat microglia and astrocytes suggesting a possible interaction between these glial cell types (Thonhoff and others 2011). Nitroxidative stress may mediate microglial toxicity to MNs; however, this appears to be specific to microglia prepared from symptomatic mutant SOD1 expressing mice, so this effect is unlikely to be a primary cause of MN degeneration (Thonhoff and others 2011).
Protein Homeostasis
Unfolded Protein Response/Endoplasmic Reticulum Stress
The endoplasmic reticulum (ER) is a cellular compartment in which post-translation protein processing, including chaperone-assisted protein folding, is carried out. When the function of the ER is perturbed, ER stress activates two adaptive pathways: (1) the unfolded protein response (UPR) to refold misfolded proteins, and (2) ER-associated degradation (ERAD) to export misfolded proteins from the ER to the ubiquitin proteasome system (UPS) for degradation. Although ER stress pathways are initially protective responses, prolonged activation can lead to pro-apoptotic consequences.
The UPR has three main pathways (the activating transcription factor 6 [ATF6], endoplasmic reticulum to nucleus signaling 1 (also known as inositol-requiring kinase 1) [IRE1], and eukaryotic translation initiation factor 2-α kinase 3 (also known as protein kinase RNA-activated (PKR)-like ER kinase) [PERK] pathways) that are all activated in spinal MNs in SOD1-ALS mouse models (Walker and Atkin 2011). This activation appears to be an early, presymptomatic, event in SOD1G93A mice, with toxic consequences for the affected cells (Saxena and others 2009). Similarly, RNA profiles of MNs derived from a SOD1+/A4V fALS patient, show activation of ER stress and the UPR, compared with non-mutant isogenic controls (Kiskinis and others 2014).
Endoplasmic reticulum stress initially increases phosphorylation of eIF2α via PERK activation, which stalls protein synthesis (anti-apoptotic ER stress pathways). However, chronic ER stress activates GADD34 which dephosphorylates eIF2α, reinitiating protein translation in a pro-apoptotic pathway (Fig. 3). Blocking the dephosphorylation of p-eIF2α, increased survival of SOD1+/A4V MNs (Kiskinis and others 2014), and this could be due to stopping the pro-apoptotic action of GADD34. However, as the basal state of SOD1+/A4V MNs was of increased p-eIF2α these results are difficult to interpret.
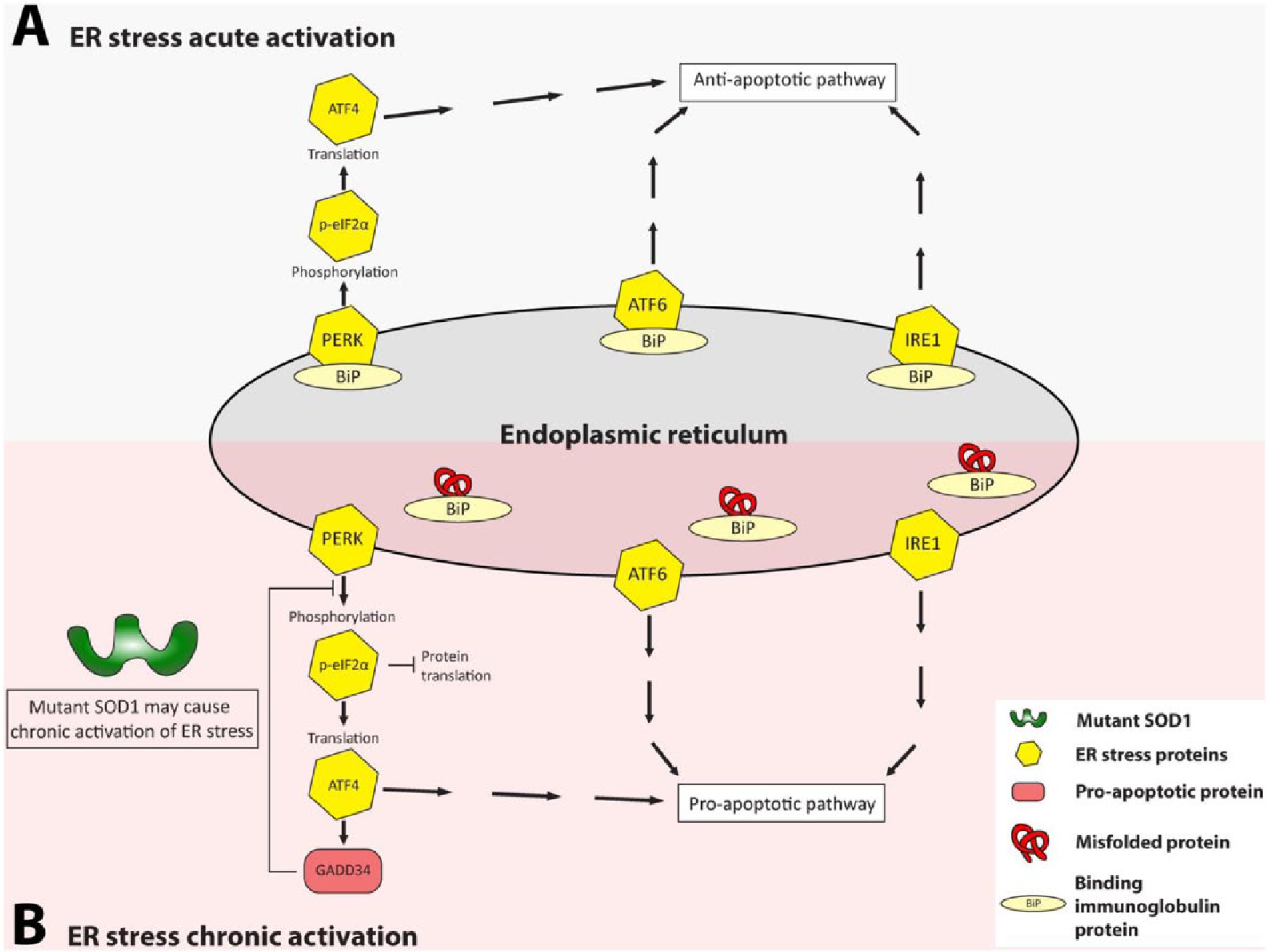
Mutant superoxide dismutase 1 (SOD1) may cause chronic activation of endoplasmic reticulum (ER) stress. Acute anti-apoptotic and chronic pro-apoptotic ER stress pathways: (A) during acute activation of ER stress the three signal transduction pathways (the PERK, ATF6, and IRE1 pathways) mediate the unfolded protein response. PERK, ATF6, and IRE1 proteins are associated with BiP (binding immunoglobulin protein). (B) However, if there is chronic activation of the ER stress PERK, ATF6, and IRE1 dissociate from BiP causing the activation of pro-apoptotic pathways. GADD34 is activated in the PERK pathway, promoting the dephosphorylation of p-elF2α and the reinitiation of protein synthesis. Chronic activation of ER stress, may mediate the unfolded protein response (UPR) and the ER-associated degradation (ERAD) mechanism.
Mutant SOD1G85R transgenic mice in which Perk is reduced (heterozygous knockout), have shortened disease duration and lifespan, perhaps because the UPR is overwhelmed by misfolded mutant SOD1, whereas SOD1G85R transgenic mice in which the effect of PERK is increased by reduction of Gadd34 (heterozygous knockout), have an ameliorated disease and prolonged lifespan (Wang and others 2011; Wang and others 2014).
Mutant SOD1 has been implicated in the direct activation of ER stress through interaction with subunits of coatomer coat protein II complex (COPII) in a transfected cell model and in mutant SOD1 transgenic mice as early as 10 days of age (Atkin and others 2014). COPII is essential for ER-Golgi transport, and expression of mutant SOD1 caused ER-Golgi transport disturbance before either the earliest markers for ER stress or aggregate formation, implicating the interaction and its effect as an early upstream event in mutant SOD1 toxicity. Disrupting ER-Golgi transport in this way results in activation of ER stress responses (Fig. 4). In cellular models, co-expression of COPII subunits with mutant SOD1 significantly reduced SOD1-induced apoptosis (Atkin and others, 2014).
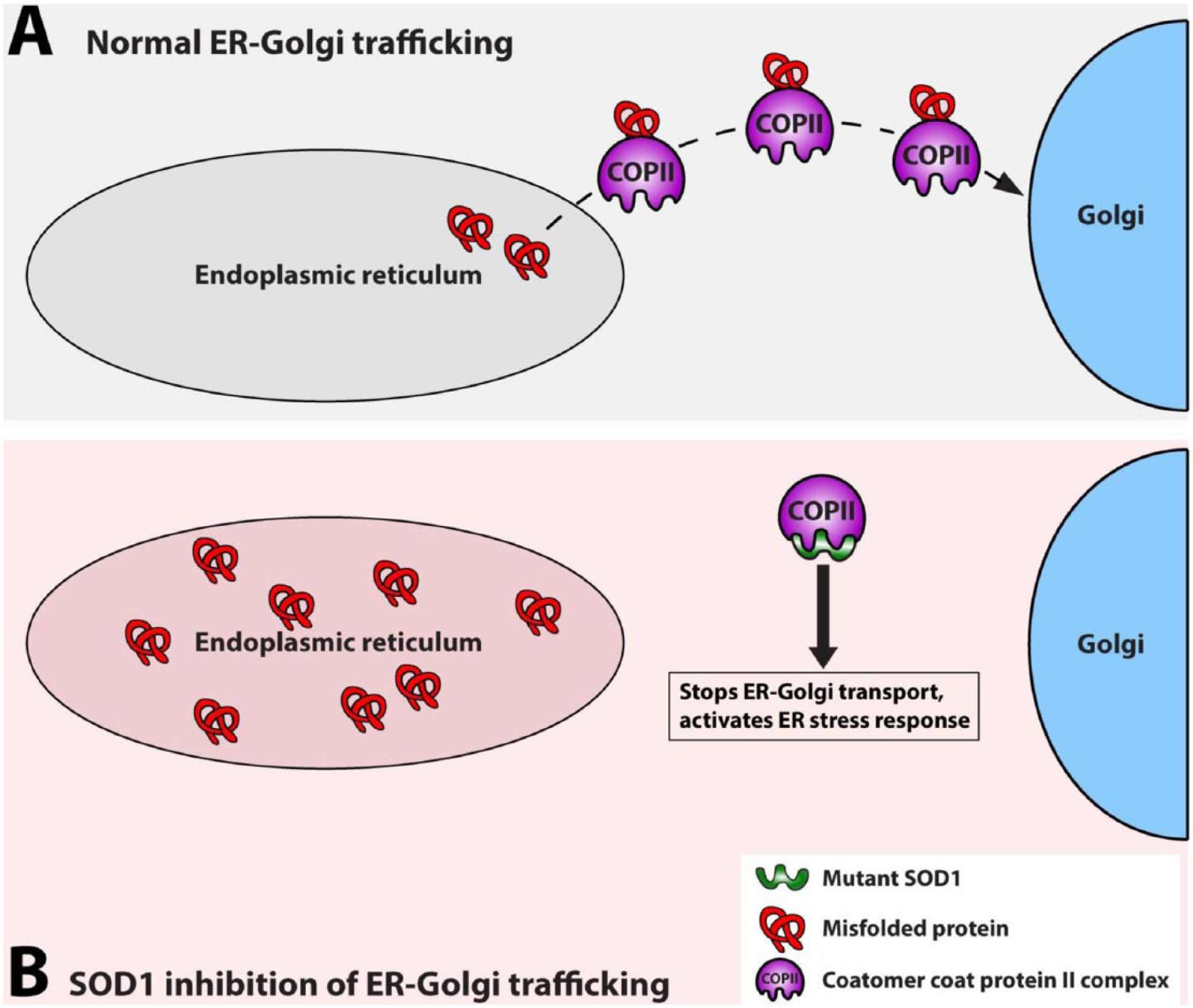
Mutant superoxide dismutase 1 (SOD1) activates endoplasmic reticulum (ER) stress by disrupting ER-Golgi trafficking through binding to coatomer coat protein II complex (COPII). (A) In normal conditions, COPII transports normal and misfolded proteins from the ER to the Golgi (only misfolded proteins are show in the figure) but (B) when mutant SOD1 is present, it interacts with a COPII subunit disrupting ER-Golgi trafficking resulting in a buildup of misfolded proteins in the ER, leading to ER stress.
SOD1 as an ER Stress Activating Zinc Sensor
Another recently proposed role for SOD1 is as a zinc sensor, activating ER stress and up-regulating zinc transporters by binding to Derlin1 in zinc-deficient conditions (Homma and others 2013). Derlin1 is an ERAD protein involved in export of misfolded proteins from the ER to the UPS. Under conditions of zinc depletion SOD1, which is a copper/zinc dependent enzyme, binds Derlin1 at a normally hidden Derlin1 binding region, and the resulting dysfunction of both Derlin1 and the ERAD process causes a build-up of misfolded protein in the ER and elicits ER stress (Fig. 5). ATF6, a transcription factor activated by ER stress, as well as up-regulating ER chaperones, also promotes transcription of the zinc transporter ZIP14, which may act to restore intracellular zinc levels (Homma and others 2013).
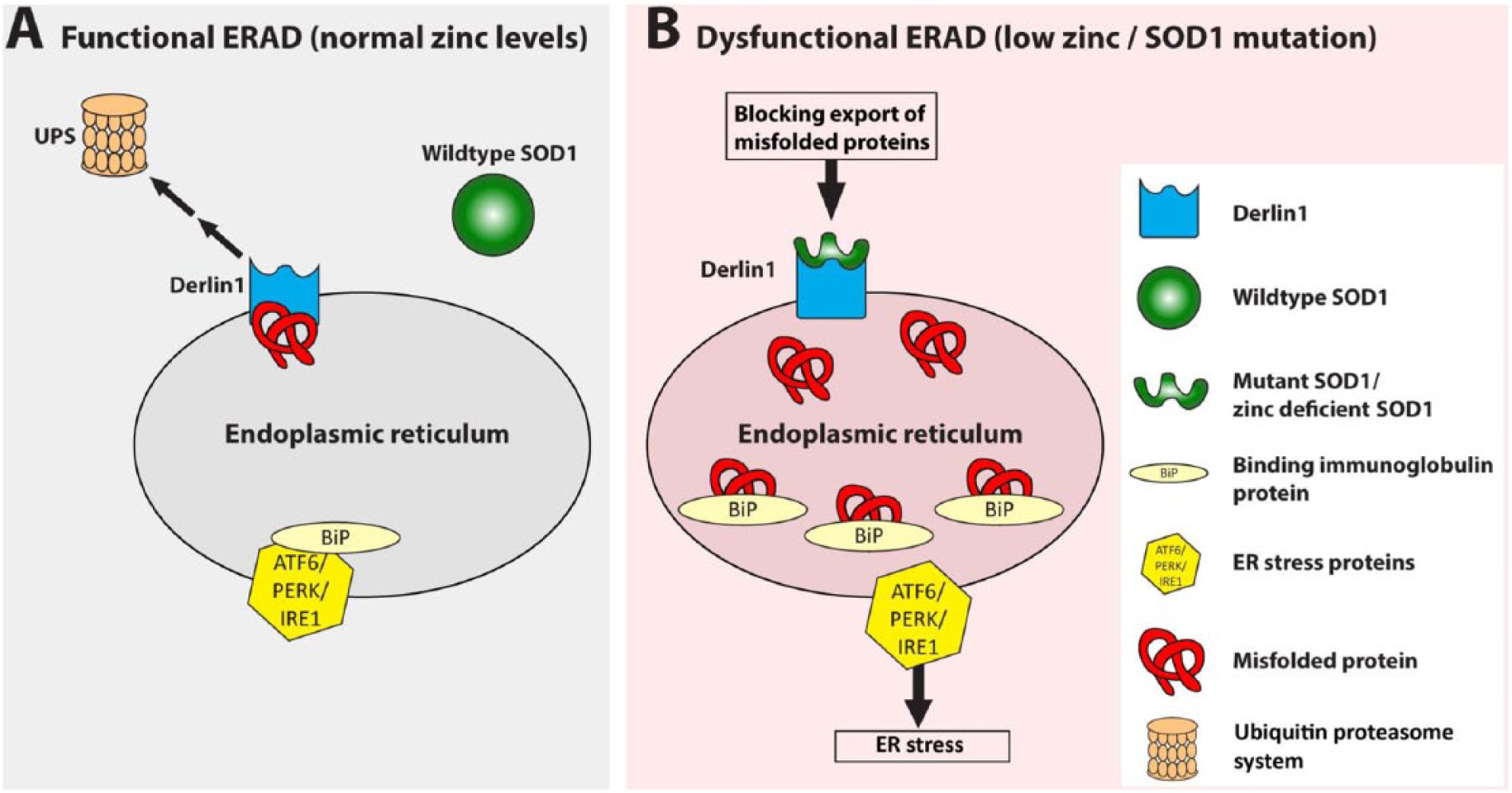
Superoxide dismutase 1 (SOD1) as an endoplasmic reticulum (ER) stress activating zinc sensor. (A) Under normal conditions wildtype SOD1 does not bind Derlin1, allowing export of misfolded proteins to the ubiquitin proteasome system (UPS). The three ER stress signal transducers PERK, ATF6, and IRE1 remain inactive and associated with binding immunoglobulin protein (BiP). (B) When mutant and/or in conditions of zinc depletion SOD1 assumes a mutant-like conformation exposing a binding site for Derlin1. The SOD1-Derlin1 interaction causes an accumulation of misfolded proteins which sequester BiP away from PERK, ATF6, and IRE1, activating the homeostatic ER stress response. Therefore, mutation of SOD1 in amyotrophic lateral sclerosis (ALS) may lead to constitutive Derlin1–SOD1 binding and ER stress activation.
SOD1 mutation in ALS may result in constitutive exposure of the Derlin1 binding region and chronic ER stress and ERAD dysfunction. Mutant SOD1 activates ER stress and the IRE1-ASK1 pro-apoptotic pathway in a Derlin1 binding-dependent manner (Nishitoh and others 2008); further, of 132 ALS associated SOD1 mutations, 124 co-immunoprecipitated with Derlin1 in transfected HEK cells (Fujisawa and others 2012). The mutants which did not co-precipitate with Derlin1 also failed to activate ER stress and were less toxic in cellular assays (Fujisawa and others 2012). In a SOD1G93A transgenic mouse model of ALS, knocking-out Ask1 improved survival and reduced MN death, showing that this pathway is also important in vivo, however, age at disease onset was unchanged (Nishitoh and others 2008).
Motor neurons may have an increased basal level of ER stress as compared with other neuronal and non-neuronal cell types; this difference might be a source of the selective vulnerability of MNs in ALS (Kiskinis and others 2014) and might also make them more susceptible to perturbation of Derlin1 function by mutant SOD1.
Autophagy
The autophagy pathway for protein clearance has been implicated as dysfunctional in sALS and fALS patients (unidentified genotypes) and transgenic mouse models of SOD1-fALS (Hetz and others 2009, and reviewed in Song and others 2012). Pharmacological and genetic manipulation of autophagy pathways in SOD1-fALS mice and Caenorhabditis elegans models has resulted in conflicting findings suggesting both protective and toxic roles for autophagy (Hetz and others 2009; Li and others 2013; Song and others 2012; Zhang and others 2011; Nassif and others 2014).
A direct role for mutant SOD1 in abnormal autophagy is suggested as, when overexpressed in a neuronal-like cell line, mutant SOD1 was found to co-precipitate with beclin 1 (BECN1), an autophagy activator, and with BCL2L1, a suppresser of BECN1 activity, whereas wildtype SOD1 only precipitated with BCL2L1 (Nassif and others 2014). The BECN1–BCL2L1 complex was weakened by co-expression with mutant SOD1, suggesting mutant SOD1 may activate autophagy by releasing BECN1 from the suppression of BCL2L1. However, the effect of wildtype SOD1, or any other co-expressed protein, on the BECN1-BCL2L1 complex was not assessed, therefore it is not possible to attribute the effect specifically to mutant SOD1 rather than increased protein expression per se. The activation of autophagy by mutant SOD1 could also be a downstream consequence of early effects on axonal transport and ER stress and ERAD (Hetz and others 2009; Zhang and others 2007).
Other Emerging Roles for SOD1
Several causative “ALS genes” encode RNA binding proteins, and this has highlighted how disruption of processes involving RNA may play a primary role in the pathogenesis of the disease. Among these proteins, TDP43 and FUS are to date the most studied (Ling and others 2014; Nassif and others 2014). However, recent findings also point to a potential role of SOD1 in nucleic acid metabolism, as below.
SOD1 as a Transcription Factor
Although this is not their best characterized role, both TDP43 and FUS have been shown to act as transcription factors and, similarly, recent evidence has shown that SOD1 can regulate transcription in response to oxidative stress stimuli by moving into the nucleus and binding to promoters to regulate the expression of oxidative resistance genes (Hu and others 2009; Tsang and others 2014).
SOD1 as an RNA Binding Protein
TDP43 (Transactive response DNA Binding protein 43kDa) and FUS (Fused in Sarcoma) are involved in mRNA splicing, miRNA biogenesis, and mRNA stabilization and transport (Ling and others 2014). Whilst evidence of a role for SOD1 in the former two is still lacking, mutant SOD1 can bind mRNAs and play a role in their stabilization (Lu and others 2007; Lu and others 2009; Chen and others 2014). Wildtype SOD1 has further been shown to interact with TDP43, suggesting a potential common action in the regulation of specific RNA stability (Volkening and others 2009).
Mutant SOD1 binds sequence elements within the 3′ UTR of VEGF (Vascular Endothelial Growth Factor) mRNA and forms complexes with other ribonucleoproteins such as TIAR and HuR. These interactions, which are specific to mutant SOD1, negatively affect levels of VEGF mRNA, which is a neuroprotective factor for motor neurons (Lu and others 2007; Lu and others 2009). Similarly, mutant SOD1 has been shown to bind the 3′ UTR of the neurofilament light chain (NFL) mRNA and negatively affect its stability. In an induce Pluripotential Stem Cell-derived cell model of ALS this reduction of NFL mRNA level mediates axonal degeneration, which could be a critical first step in ALS (Chen and others 2014).
SOD1 as a Signaling Molecule
SOD1 and Cell Metabolic State Signaling
Using yeast and human cell lines Reddi and Culotta (2013) identified a new role in cellular metabolism for SOD1: to integrate signals from oxygen and glucose in order to repress respiration within cells. In the mechanism proposed SOD1 binds the casein kinase gamma homologues Yck1p and Yck2p preventing their degradation via its enzymatic activity (Fig. 6). This results in repression of aerobic respiration and promotion of aerobic fermentation. Without SOD1, Yck1p and Yck2p are degraded, resulting in aerobic respiration. This finding gives insight into how rapidly proliferating cells may favor anaerobic glycolysis. Although effects in neurons remain to be determined the authors note the link between this signaling pathway and responses to hypoxia, a toxic state for motor neurons (Reddi and Culotta 2013). It is also interesting to note that a de novo mutation in casein kinase 1 gamma 3 (CSNK1G3) was recently identified as a potential ALS risk factor (Chesi and others 2014) and, in a yeast screen YCK2, a homologue of the human casein kinase 1 gamma 2, was identified as an enhancer of TDP43 toxicity (Kim and others 2014), offering hints that the putative interaction between SOD1 and casein kinase 1 gamma could be relevant to ALS.
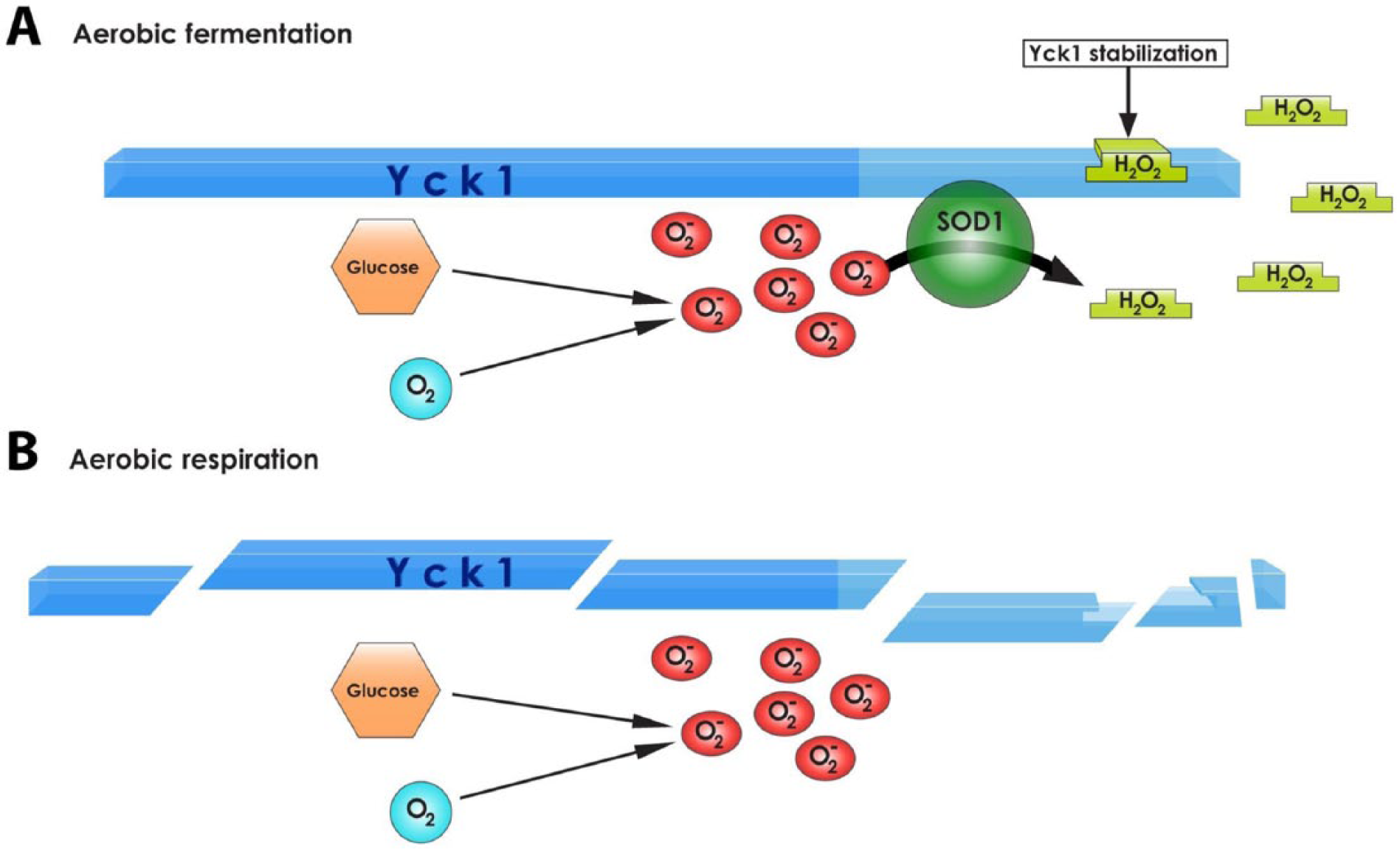
The role of superoxide dismutase (SOD1) in cellular metabolism, in yeast. (A) In an environment rich in glucose and oxygen SOD1 binds to the C-terminal degron of Yck1p and prevents its degradation, producing hydrogen peroxide that stabilizes the complex, so enabling the cells to utilise aerobic fermentation. (B) When SOD1 is not present Yck1p is degraded causing the cells to undergo aerobic respiration.
Conclusions and Outstanding Questions
In ALS, SOD1 takes on a toxic unknown gain of function. However, as with many proteins involved in neurodegenerative disease, it is still not clear what the normal functions are for SOD1, and which cellular pathways rely on this protein. We know that it is abundant and ubiquitous, but protein levels appear to be far above what would be required solely for its role as an antioxidant in the cytosol.
New functions are now coming to light, and intriguingly—given the roles of other causative ALS genes—SOD1 is involved in RNA metabolism, including acting as a nuclear transcription factor, and in metabolic signaling. Almost certainly there is much more to be discovered about SOD1 function in the normal situation, as well as in ALS, where there are interesting links and converging themes involving excitotoxicity (AMPA receptors, sodium channel activation) and ER stress, as well as cell specific effects from astrocytes and microglia.
Study of these functions present new opportunities for badly needed therapeutics to modulate disease progression in SOD1-fALS, and possibly even sALS, perhaps in combination with anti-sense oligonucleotide therapies (Musarò 2013; Yang and others 2013). However, until we have a considerably better understanding of SOD1 functions, much about this small protein remains enigmatic.
Footnotes
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: The authors have been supported by the Motor Neuron Disease Association (RAS, PF, EMCF), the Thierry Latran Foundation (PF, EMCF), the UK Medical Research Council (RKAB-S, PF, EMCF) and the NIHR-UCLH Biomedical Research Centre (PF).