
Abstract
The acquisition of genetic- and epigenetic-abnormalities during transformation has been recognized as the two fundamental factors that lead to tumorigenesis and determine the aggressive biology of tumor cells. However, there is a regularity that tumors derived from less-differentiated normal origin cells (NOCs) usually have a higher risk of vascular involvement, lymphatic and distant metastasis, which can be observed in both lymphohematopoietic malignancies and somatic cancers. Obviously, the hypothesis of genetic- and epigenetic-abnormalities is not sufficient to explain how the linear relationship between the cellular origin and the biological behavior of tumors is formed, because the cell origin of tumor is an independent factor related to tumor biology. In a given system, tumors can originate from multiple cell types, and tumor-initiating cells (TICs) can be mapped to different differentiation hierarchies of normal stem cells, suggesting that the heterogeneity of the origin of TICs is not completely chaotic. TIC’s epigenome includes not only genetic- and epigenetic-abnormalities, but also established epigenetic status of genes inherited from NOCs. In reviewing previous studies, we found much evidence supporting that the status of many tumor-related “epigenetic abnormalities” in TICs is consistent with that of the corresponding NOC of the same differentiation hierarchy, suggesting that they may not be true epigenetic abnormalities. So, we speculate that the established statuses of genes that control NOC’s migration, adhesion and colonization capabilities, cell-cycle quiescence, expression of drug transporters, induction of mesenchymal formation, overexpression of telomerase, and preference for glycolysis can be inherited to TICs through epigenetic memory and be manifested as their aggressive biology. TICs of different origins can maintain different degrees of innate stemness from NOC, which may explain why malignancies with stem cell phenotypes are usually more aggressive.
Introduction
Currently, there are two dimensions for understanding tumorigenesis and tumor biology. The first one is genetic abnormality that is defined as any alteration in the DNA sequence that causes disease. In the past 50 years, a large number of tumor-related genetic variants were identified, which has brought about tremendous changes in the cognition of tumorigenesis and treatment. The second one is epigenetic abnormality, also called as epigenetic mutation (epimutation), that is defined as abnormal transcriptional repression of active genes and/or abnormal activation of usually repressed genes caused by errors in epigenetic gene repression.1,2 Later, it was further extended as epigenetic plasticity, referring to a “plastic” state that allows stochastic oncogene activation. 3 Functionally, epigenetic abnormalities are thought to be equivalent to genetic mutations that lead to neoplastic transformation. In addition, some researchers have proposed that, just like genetic changes, epigenetic changes in tumors are gradually accumulated in the process of transformation and evolution. 4
Most tumors are clonal. That is, a normal cell gains the advantage of clonal proliferation by accumulating adequate driver mutations, thus becomes a tumor-initiating cell (TIC). All tumor parenchymal cells that make up the tumor mass are descendants of this TIC. 5 Cell division is probably the most well-known way of introducing mutations, accumulating mutations, and passing mutations on to offspring. During selection under stress conditions, cells at any stage of differentiation may accumulate enough driver mutations to transform. Although mutations disturb the epigenome of normal cells and contribute to the formation of TIC’s epigenome, TIC and its counterpart normal cells of the same differentiation hierarchy may have greatest degree of epigenetic similarity. In this work, we define the normal cells at the same hierarchy of differentiation as TIC as normal origin cells (NOCs).
Interestingly, there is a phenomenon that tumors derived from less-differentiated NOCs or TSCs usually have a higher risk of vascular involvement, lymphatic and distant metastasis. This regularity is first seen in lymphohematopoietic malignancies. For example, the biological behavior of leukemia stem cells and hematopoietic malignancies with hematopoietic stem cell phenotypes is generally more aggressive. 6 The biological behavior of lymphoma or leukemia derived from mature cell types, such as B-cell chronic lymphocytic leukemia/small lymphocytic lymphoma (B-CLL/SLL), mantle cell lymphoma, plasma cell lymphoma, and follicular lymphoma, is relatively inert.7,8 In recent years, the influence of cellular origin on tumor biology has been observed in a wide range of somatic tumors.9-11 In a given system, cells of different lineages and stages of differentiation have distinct biomarker expression profiles, which have been formed over a long period of species evolution and are strictly programmed. Accordingly, the tracing of tumors’ cellular origins is achieved based on the conserved expression profiles of these biomarkers in tumors. Given that the cellular origin of tumors is an independent indicator that associated with tumor biology, we speculate that there may be other mechanism that contributes to the linear relationship between cellular origin and the biological behavior of tumors.
Limitations in Understanding of the Cellular Origin of Tumors May Lead to Systemic Deficiencies in the Identification of Tumor-Associated Epigenetic Abnormalities
Main Models of Tumorigenesis
Currently, there are two main hypotheses about the origin of a TIC, and the principles of genetic variation and clonal selection are still the foundations of the hypotheses. The first is the stochastic model (or clonal evolution model), which proposes that as long as adequate driver mutations are accumulated, any proliferating cell may gain the advantage of clonal proliferation and become a TIC. The second is the tumor stem cell (TSC) model, which proposes that a normal cell escapes regulation and gives rise to a stem cell-like counterpart, a TSC. 12 Some researchers have suggested that TSCs are just the transformed products of normal stem cells or progenitor cells. 13 In addition, many scholars believe that TSCs may derive from a few tumor-initiating cells, which are reprogrammed differentiated cells after acquiring initial cancer-causing mutations. 14 Recently, some excellent reviews have summarized the historical developments and new advances in tumorigenesis models.15,16 These models incorporated non-genetic determinants into stochastic model and TSC models and highlighted the tumorigenic epigenetic changes via cellular reprogramming or microenvironmental stress. All of these models are reasonable hypotheses to describe tumorigenesis and tumor heterogeneity, but each model is insufficient to explain how the linear relationship between the cellular origin and the biological behavior of tumors are formed.
The Multi-Lineage Cellular Origin of TIC in a Given System
Lineage analysis studies for cancer heterogeneity.

By analyzing the cumulative process of driver mutations in cell differentiation and self-renewal, a model was developed to describe the heterogeneity of the origin of TSCs (Figure 1A). Cell division is the cytological basis for genetic laws and biological variation. Through asymmetric or symmetric division, two basic modes of cell division, the fate of stem cells is determined towards differentiation or self-renewal.
24
In either mode, genetic abnormalities in DNA can be accumulated and transmitted to progeny cells. Stem cells have the highest potential for proliferation and a much longer life span compared with their progeny and therefore have a greater opportunity to accumulate mutations through self-renewal or differentiation.
25
However, mature cells can accumulate mutations and pass them on to their descendants through only limited rounds of self-renewal. Generally, the surveillance mechanisms of the human body can identify the illegitimate changes.
26
In normal conditions, if the surveillance system identifies genetic alterations, the repair mechanisms, such as p53-dependent pathway, may correct the errors and the cell will survive. If the alterations cannot be corrected, cells with a malignant potential (precancerous cells) can be removed by apoptosis mechanisms.
27
In other cases, if the body cannot recognize and repair the illegitimate changes, they will be carried by precancerous cells and be passed to all offspring through self-renewal and differentiation. As long as these mutation carrying cells do not lose their ability to differentiate, they may enter the apoptotic program. Otherwise, they can survive. Under selective pressures, proliferating cells of any differentiation stage can be at risk of introducing and accumulating mutations, it is reasonable to infer that the proliferating cells at any differentiation stage can accumulate adequate driver mutations to achieve the advantage of clonal proliferation. Thus, not only pluripotent stem cells, blasts/progenitor cells, and lineage-committed progenitor cells,
28
but also mature cells
29
can be qualified as candidates for the origin of TSCs. Given the common feature of clonal proliferation of benign and malignant tumors, this model can be used to explain the heterogeneity of cellular origins of both in a given system. A. In a given system, TICs of different tumors or their subtypes can be mapped to different differentiation hierarchies of normal stem cells. Clonal proliferation is characteristic of both benign and malignant tumors. Before malignant transformation, normal cells usually undergo a precancerous phase. Here, we hypothesized that when a normal cell accumulates an average of three driver mutations, it becomes a precancerous cell or a clonal proliferating benign TIC. Further, when an average of four drive mutations is accumulated, a malignant transformation occurs. By differentiation and self-renewal, any proliferating cells can introduce driver mutations to become precancerous cells. Precancerous cells carrying a small number of driver mutations may be histologically indistinguishable from normal cells, but they may exhibit atypia as the driver mutations are further accumulated. However, precancerous cells did not gain the advantage of clonal proliferation and eventually entered the apoptotic program. In developing and adult organs, the differentiation hierarchies of parenchymal cells exist. Through self-renewal and/or differentiation, proliferating cells at any stage of differentiation can accumulate adequate drive mutations to become TICs. Therefore, TICs can be mapped to different differentiation hierarchies of normal stem cells in a given system. B. The stemness of TIC of different origin may be parallel to that of their corresponding NOC. Normal stem cells are characterized by their stemness properties. As normal stem cells differentiate towards maturity, their stemness gradually decreases and eventually disappears. Although stochastic genetic or epigenetic mutations drive transformation, they cannot cause earth-shaking changes in the epigenome of TIC. The physiological characteristics of NOC, especially its stemness, can be inherited (perhaps not entirely) to its corresponding TIC and are presented by the aggressive biology of TIC. TIC of different origins can inherit different degrees of stemness from NOC, which may explain why a malignancy derived from a less differentiated NOC usually have a higher risk of vascular involvement, lymphatic and distant metastasis.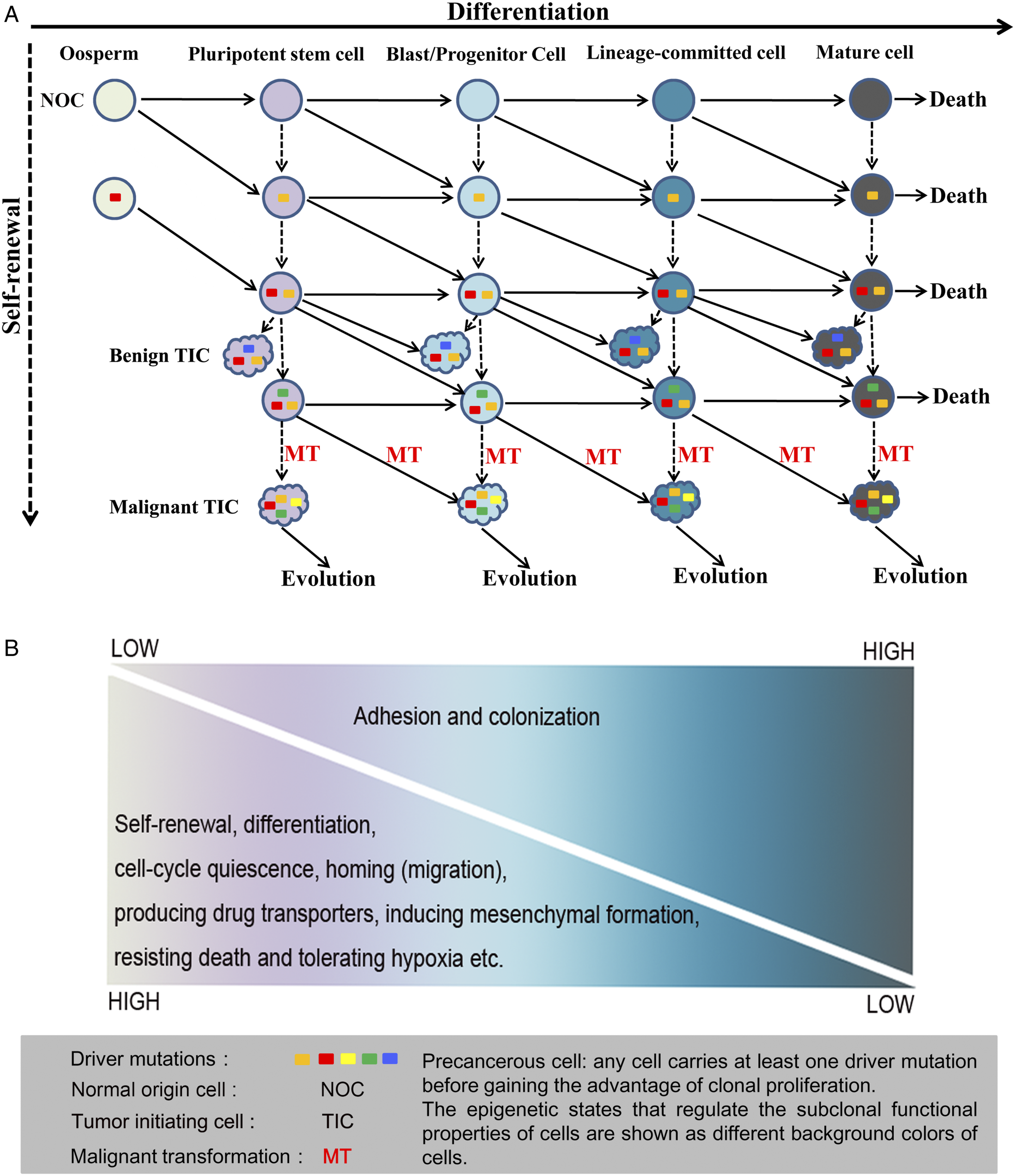
For both TIC and its corresponding NOC, all the biological characteristics are determined by their respective epigenomes, that is, all epigenetic programs that regulate the expression of all genes within the genome. 30 TIC’s epigenome includes not only epigenetic-abnormalities, secondary epigenetic-abnormalities (epigenetic aberrance due to genetic changes), 31 but also the established epigenetic status of genes inherited from NOC. Of which, secondary epigenetic-abnormalities are the most well-known aspect that contributes to the malignant phenotype of tumors. While thousands of genetic alterations have been identified in different tumors, several studies have revealed that most of the genetic changes in tumors are passenger mutations, with only a few driver mutations (four on averages) sufficient to convert a normal cell into a cancer cell.32,33 Clearly, limited genetic variation is not sufficient to cause the great heterogeneity within a given tumor, let alone explain the laws behind it. 34 Naturally, the relationship between epigenetic factors and tumorigenesis and biological behavior of tumor cells has attracted researchers' attention and become a research hotspot.
Which Normal Cell Should Be Used as a Control to Identify the Tumor-Associated Epigenetic Abnormality?
So far, there is no consensus on explaining how epigenetic abnormalities occur in tumors. Cell’s stochastic epigenetic regulatory changes and response to tumorigenic factors or microenvironmental changes are two major hypotheses.35,36 In practice, the identification of epigenetic abnormalities in tumors was commonly based on the differences in epigenetic status of certain gene between tumor bulks/cells and normal control, mostly tissue bulks from the excised organs.37,38 Normal tissues, non-tumor tissues under the background of chronic diseases, and tumor tissues all have parenchymal and mesenchymal heterogeneous composition, 39 which suggests that the method of identifying epigenetic abnormalities of tumors with tissue masses has systemic defects. Ideally, the identification of epigenetic abnormalities in certain tumors or tumor subtypes should be based on differences in epigenetic status of genes between TIC and corresponding NOC, rather than on normal or pericancerous tissue bulks. Otherwise, it will not be possible to reliably distinguish whether the differential epigenetic event is the innate epigenetic state of the gene inherited from the NOC or the real epigenetic abnormality.
Epigenetic Similarities Between Tumors and Their Corresponding NOCs
The definition of TIC emphasizes the cellular origin of tumors, while the definition of TSC emphasizes the cells with the characteristics of stem cells in the tumor mass. In some early and even current studies, researchers did not make a strict distinction between TSCs and TICs. 40 We now know that although cells with stem cell characteristics may not represent the real TICs, comparing the epigenetic status of genes of interest between TSCs and the corresponding normal stem cells can still help reveal the epigenetic regulation of the biological behavior of tumor cells. Known epigenetic events are of 3 types: methylation of DNA and RNA, histone modification (acetylation, methylation, and phosphorylation), and the expression of non-coding RNA. 41 The epigenetic similarity between TSC and NOC may provide some clues for our understanding of the formation of tumor biology.
DNA Methylation
In mammals, DNA methylation is the main form of epigenetic regulation that controls embryonic development, cell differentiation, and lineage specification. Stem cells and differentiated cells have distinct global DNA methylation profiles. Compared to the differentiated cells, the global DNA of the stem cells is usually in a hypomethylated state. With differentiation, the cell’s global DNA methylation level increases.42,43 Interestingly, although DNA hypomethylation can be observed in various tumors, global DNA hypomethylation is preferentially distributed in high-grade cancers and in tumors with a stem cell phenotype.44-46 In liver, global DNA hypomethylation is significantly detected in HCCs with stem cell phenotypes47,48 and hepatoblastoma 49 than in well-differentiated HCC and adjacent tissues. In well-differentiated thyroid neoplasms, global hypermethylation, rather than global hypomethylation was found, which is consistent with that of normal mature thyroid epithelium. 50 In the lymphopoietic system, Kulis and his colleagues found extensive overlap of methylated regions between multiple B cell neoplasms and their corresponding NOCs. 51 In other systems, preference distribution of the global DNA hypomethylation in tumors with stem/progenitor cell characteristics was also observed.52-54
Coupled with global DNA hypomethylation, another hallmark of many tumors is local hypermethylation, which mainly occurs on the promoter CpG islands of tumor suppressor genes. Many scholars have proposed that methylation of promoters of these genes was stochastically acquired during tumorigenesis.55,56 With the help of genome-wide DNA methylation scans, many researchers have evaluated the alterations of the DNA methylation landscape in various human tumors. In their studies, although considerable differential CpG island methylation was found, only a small proportion of them were candidate drivers for tumorigenesis.57-61 Furthermore, results from several independent groups have shown that a large number of TSCs-specific methylation and their occurrence sites overlap with those of normal stem cells.55,62-65 A study revealed that abnormal methylation patterns in prostate cancer were associated with dynamic patterns of DNA methylation from the normal basal prostate epithelial cell differentiation towards luminal cell. 66 This makes people wonder whether the abnormal methylation of prostate cancer is formed via the epigenetic inheritance from the basal cell during the malignant transformation. Di Domenico analyzed genome-wide DNA methylation data of 125 Pancreatic Neuroendocrine Tumors (PanNETs). Based on epigenetic similarities, they found that PanNETs cluster in pancreatic α-like cell, β-like cell, and intermediate tumors. 67 Shen observed that DNA methylation patterns of embryonal carcinoma (EC) resembles that of embryonic stem cells, while non-EC non-seminomatous germ cell tumors adopt DNA methylation patterns resembling somatic and extraembryonal lineages. 68 Smith and colleagues found that small cell neuroendocrine carcinomas of lung, prostate, and bladder are enriched for a transcriptional signature shared by epithelial adult stem cells. Further, DNA methyltransferase expression correlated with adult stem cell signature status was enriched in these small cell neuroendocrine carcinomas, and a highly consistent DNA methylation profile across the carcinomas was found. Most interestingly, adult stem cell signatures associated with the small cell neuroendocrine carcinomas were not significantly affected by other molecular signatures. 69 Recently, Giacopelli analyzed genome-wide DNA methylation in 649 acute myeloid leukemia (AML) patients. They revealed that epitypes of AML subtypes showed developmental arrest at discrete stages of myeloid differentiation, revealing epitypes that retain arrested hematopoietic stem-cell-like phenotypes. Further, they found that patients in epitypes with stem-cell-like methylation features showed poor overall survival along with up-regulated stem cell gene expression signatures. 70 In many tumors or TSCs, there are signs of de novo DNA methylation, involving essential DNA methyltransferase (Dnmt), especially Dnmt3a and Dnmt3b. Professor Meissner’s research shows that DNA methylation profiles of 15 cancer types is consistent with de novo DNA methylation of extraembryonic ectoderm, and are regulated by Dnmt3a and Dnmt3b, indicating that they can use similar mechanisms. 55
In addition to CpG islands, there are a large number of DNA methylation changes outside of CpG islands, such as the methylation of “CpG island shores” in tumors. CpG island shore methylation is strongly related to gene expression, and it is highly conserved in mouse cells, discriminating tissue types regardless of species of origin. Irizarry and colleagues found a broad overlap between colon cancer-related methylation and stem cell differentiation-related methylation in CpG island shores. 71 Doi and colleagues reported that their target methylated regions in CpG island shores for pluripotent stem cells, embryonic stem cells, and fibroblasts differentiation largely overlap those of the previously reported aberrant methylation in cancers. 72 Based on the DNA methylation profiles and CpG sites, Tang and colleagues develop a classifier that can accurately identify the primary site of tumor. 73
Histone Modification
A histone modification is a covalent post-translational modification to histone proteins that can impact gene expression by altering chromatin structure or recruiting histone modifiers. 74 Histone modification includes methylation, phosphorylation, acetylation, ubiquitylation, and sumoylation of histone proteins of which histone methylation is one of the most important modifications associated with either transcriptional repression or activation. Activating (H3K4me3) and repressive (H3K27me3) histone methylations are representative events for the transcription and repression of gene expression, respectively. 75 Concomitant H3K4me3 (activating) and H3K27me3 (repressive) methylation patterns mark untranscribed lineage-specific gene loci, termed bivalent domains. Bivalent domains are usually found in embryonic stem cells and determine whether a cell will remain unspecified or will eventually differentiate.71,76-78 What is striking is the bivalent domains were detected in glioblastoma stem cells, 79 ovarian TSC-like side-population cells, 80 and stem cell-like prostate cancer cells, 81 rather than in corresponding normal astrocytes, non-side-population ovarian cancer cells, and non-stem-like prostate cancer cells, respectively. Histone methylation can occur at various sites in histone proteins, primarily on lysine and arginine residues. Histone methyltransferases catalyze histone methylations that are the important epigenetic marks regulating gene expression and cell fate. Jambhekar et al. well summarized the histone methyltransferases that regulate the histone methylation in animal development. Up-regulation of SET1, MLL, SET7/9, and SymD3 for H3K4 methylation, and EZH1, EZH2, or G9a for H3K27 methylation that helps establish bivalent chromatin domains in early embryonic systems can be observed in various TSCs. 82
Non-coding RNAs
Non-coding RNAs (ncRNAs) are functional RNA molecules that are not translated into proteins. However, ncRNAs play important roles in the regulation of gene expression. In numerous types of embryonic and adult stem cells and progenitors, the expression of ncRNAs is often tissue- or even cell type-specific, emphasizing their involvement in defining space, time, and developmental stages in gene regulation.83,84 Based on the length, ncRNAs can be divided into two major classes, small and long ncRNAs (lncRNAs). Small ncRNAs are further divided into microRNAs (miRNAs), short interfering RNAs (siRNAs), and piwi-interacting RNAs (piRNAs). Here, we briefly review the impact of miRNAs and lncRNAs on tumorigenesis.
Tordonato had compared the transcription of microRNAs between breast stem cells/progenitor cells and breast TSCs. They found that the expression of Let-7, miR-200 family, miR-22, miR-205 was consistent between normal stem cells and TSCs. 84 Divisato summarized the expression of microRNAs in various other TSCs and embryonic stem cells (ESCs). They found that in addition to the microRNAs mentioned above, epigenetic status of other microRNAs such as miR-451, miR-21, miR-29, miR-320, miR-30, miR-122, miR-155, miR-302, C19MC miRNA cluster, Mir-23a-24-27a cluster, and MiR-125a/b family between ESCs and TSCs/cancer progenitor cells of colorectal cancer, breast cancer, follicular thyroid tumor, lung adenocarcinoma, multiple myeloma, ovarian teratocarcinoma, glioma, gastric cancer, retinoblastoma, human non-small-cell lung cancer, hepatocellular carcinoma, and undifferentiated embryonal sarcoma of the liver was consistent. 85 So far, comparative study of lncRNA between normal stem cell and TSCs is just emerging, the consistence of the epigenetic status of HOTAIR and Linc-ROR between TSCs of a variety of human tumors and ESCs/adult stem cells was revealed.84-86 Differences in non-coding RNAs expression profiles between TSCs and non-TSCs have led many investigators to associate them with the conversion of epigenetic status from normal stem cells to differentiated cells.87-90 All these evidences prompt us to consider that the expression of these key ncRNAs in TSCs are not stochastic epigenetic events, but the epigenetic inheritance from NOCs.
Key Genes and Signaling Pathways
Comparison of consistency of epigenetic status of important pathways and genes between TSC and putative NOC.

Hippo signaling has been a field of very active research in recent years. Abnormal activation of YAP (Yes-associated protein), the main effector molecule of Hippo pathway, is closely related to the occurrence of various tumors. Normally, YAP activation plays an important role in promoting cell proliferation and maintaining the stemness of stem cells and progenitor cells. 111 YAP was not expressed in pericancerous tissues and mature cells. 112 Although YAP is overexpressed or enriched in the nucleus of tumor cells, especially TSCs, such as hepatocellular carcinoma, gastric cancer, colon cancer, prostate cancer, and ovarian cancer,111,113 YAP is low or not expressed in well-differentiated tumors.114-116 These evidences suggest that YAP gene can be activated not only by genetic mutation, but also by epigenetic inheritance from NOC in tumors. Further, in vitro, YAP has been shown to promote dedifferentiation of mature hepatocytes into progenitors, 117 which can imply that YAP activation, whether through genetic mutation or epigenetic inheritance, may be equivalent in determining the biological behavior of TIC.
Similarities in Biological Behavior Between TICs and Their Corresponding NOCs
The epigenetic events of cells will eventually be transformed into specific biological functions. The exploration of cellular biology and its conversion in the process of stem cell differentiation can help us understand how the physiological function of NOC manifests as the aggressive biological behavior of TIC. Figure 1B shows the gradual decrease in stemness and the increase in adhesion and colonization ability of stem cells during the differentiation, which may reflect the stemness characteristics of TICs of different cellular origins. We will elaborate below.
Cell Dynamics
The biological characteristics of normal cells are closely related to their differentiation status. Along the direction of stem cell differentiation, stemness of cells successively decreases and finally disappears in mature cells. In terms of cell dynamics, it is mainly manifested as increased cell adhesion and colonization ability and decreased cell proliferation and migration ability. 118 Cell migration capacity is a prerequisite for tumor invasion and metastasis. Previously, some scholars have noted that tumor invasion and metastasis may be associated with stem cell homing.119,120 Stem cell homing is a controlled recruitment of stem cells that leads to trans-endothelial and directional migration, which is strictly regulated by well-directed epigenetic programs in organism development, tissue regeneration and repair. 121 During differentiation, epigenetic reprogramming of stem cell is accompanied by the conversion of cellular biology. With differentiation until maturity, the epigenetic programs that drive stem cell homing are shut down, and the cells eventually lose their ability to migrate and then colonize. For example, in the context of chronic cholecystitis, stem cells in the biliary system proliferate to form new glands to compensate for the loss of the epithelial layer. 122 These new glands can appear in or through the muscle layer to form the Rokitansky-Aschoff sinuses, which may be misdiagnosed as cancerous infiltrations. 123 The formation of the Rokitansky-Aschoff sinuses is the result of tissue regeneration and repair after bile duct epithelium injury and is a typical biological manifestation of biliary stem cell activation, differentiation, migration, and colonization. Well-programmed cell kinetics can contribute to embryonic development and tissue regeneration and repair, while unconstrained cell kinetics can lead to damage of tissues and organs, especially in cancers. Cholangiocarcinomas are malignancies characterized by intensive local invasion and high incidence of distant metastasis. These tumors often occur in the context of repeated damage and repair of epithelial cells caused by chronic inflammation or biliary obstruction. Cholangiocarcinoma is a heterogeneous tumor of multi-lineage origin and some of its subtypes can express stem cell biomarker CD133. These CD133 positive cholangiocarcinomas showed significantly higher rates of nodal metastasis and positive surgical margin status. In vitro, CD133 positive tumor cells had a higher invasive ability compared with CD133 negative cells. 124 In another work, hypermethylation of multiple CpG sites at genes associated with the stem cell phenotype in some highly aggressive cholangiocarcinomas was observed. 125 Zarco et al summarized findings on the mechanisms of cellular migration that overlap between neural progenitor cells (NPCs) and brain tumor stem cells (BTSCs). They found that signal pathways (JNK, PKA/Akt, PI3K/AKT/MAPK, CXCL12/CXCR4, Slit/Robo) and proteins (semaphorins, pleiotrophin, integrins, NCAM, cadherins, matrix metalloproteinases) that participate NPCs and BTSCs migration are highly similar. 126 Placental trophoblast cells are highly invasive, able to migrate and invade the uterine wall and vasculature without being rejected by the immune system, which resembles the biological behavior of choriocarcinoma. 127 These evidences lead us to speculate that TSC may be harnessing normal stem cell mechanisms and thus being endowed with greater invasiveness.
Resistance to Chemicals and Cell Death, Induction of Angiogenesis, Promotion Inflammation, Avoiding Immune Destruction, and Maintaining the Quiescent States
Many key characteristics of normal stem cells are often reported in tumors, especially in those tumors with stem cell phenotypes or TSCs. For example, like normal stem cells, cells of many types of tumors can express drug transporters to resist chemical toxicity.128-130 As normal stem cells, TSCs in chronic myeloid leukemia and solid tumors can remain quiescent and resistant to drug damage. 41 In stem cells and many types of tumor cells, telomerase is activated to maintain the cell’s ability to continue dividing and to resist cell death.131,132 Like normal stem cells, cells of many types of tumors can induce angiogenesis.133,134 Although neoplastic angiogenesis is conspicuously different from angiogenesis in normal conditions, in the past decade, many studies showed that some genes and pathways were involved in the neoplastic angiogenesis of gliomas, breast cancer, melanoma, hepatocellular carcinoma, ovarian cancer, mesothelial sarcomas, etc. are very similar to that of their respective normal stem.135,136 In vitro, purified TSCs also showed stronger ability of inducing angiogenesis than non-TSCs. 137 Normal mature somatic cells do not recruit inflammatory cells except for pathological conditions such as autoimmune diseases and tissue damage, as well as cellular senescence. However, in embryonic development and organogenesis, immunocytes can be recruited by almost all types of stem cells and are closely related to stem cell renewal, differentiation, proliferation, migration, angiogenesis induction, and protecting stem cells against immune clearance.138-140 Cancer-related inflammation is the recruitment of immunocytes to the tumor microenvironment. These immunocytes have many functions such as promotion of tumor growth, immunosuppression, angiogenesis, and metastasis. 141 Previous studies have suggested that tumor-related inflammation may be a borrowing of a similar mechanism from normal stem cells by TSC to promote angiogenesis, metastasis and escape immune clearance.142,143
Energy Metabolism
The energy supply of normal human cells usually comes in two forms, namely glycolysis and oxidative phosphorylation. Most of the quiescent stem cell populations rely on glycolysis to provide ATP. With the differentiation of stem cells, energy metabolism shifts toward oxidative phosphorylation to support the increasing energy demands. 144 Both types of energy supply can be detected in tumor cells. Depending on tumor type, degree of differentiation, or cellular origin, the type of energy supply that tumor cells prefer to use varies. In some well-differentiated tumors,145,146 energy is mainly obtained through oxidative phosphorylation, while in tumors with stem cell phenotype or poor-differentiated cancers, such as low differentiated prostate cancer, glioblastoma, triple-negative breast cancer, and colorectal cancer, energy is mainly generated through glycolysis.147-150 Embryonal carcinoma can be induced by retinoic acid to differentiate toward neurons and ectoderm lineages. In differentiated tumor cells, a metabolic transition from glycolysis to oxidative phosphorylation was observed. 151
As discussed above, a large amount of evidence to demonstrate the similarity in many core biological properties and epigenetic regulation between multiple tumors and their putative NOCs has been obtained. Collectively, we suggest that the established epigenetic states of genes in NOCs can be inherited to TICs. Many neoplastic biology of TIC may be a translation of innate epigenetics inherited from NOC rather than random epigenetic abnormalities. From pluripotent stem cells, blasts/progenitor cells, lineage committed stem cells to mature cells, the stemness of the cells decreases sequentially. 152 Parallel to this, TICs of different origins can inherit different degrees of stemness that are determined by the established epigenetic states of genes of NOCs, which may explain why malignancies that derived from less-differentiated NOCs usually have a higher risk of vascular involvement, lymphatic and distant metastasis. In vitro experiments have shown that TICs with stem cell phenotypes have the ability to differentiate, and their differentiated progeny generally have reduced or disappeared tumorigenic and invasive capabilities.153,154 Clearly, this phenomenon cannot be explained by random genetic- or epigenetic-abnormalities, but is similar to the conversion of physiological functions that occurs during normal stem cell differentiation.
Innate Post-Transcriptional Regulation of NOC May Be Transferred to TIC via Epigenetic Memory
Propagation of the chromatin landscape across cell divisions is central to epigenetic memory of cells. Understanding the intergenerational transmission of chromatin status in self-renewing cells might shed some light on how the epigenetics of NOC is passed on to TIC. In the nucleus, DNA wrapped around histones form nucleosomes that allow for compaction of DNA and assist in gene regulation. There are many post-translational modifications on histones, including phosphorylation, acetylation, methylation, ubiquitination, and ADP-ribosomal modification, with cell-specific sites, degrees, and patterns. 155 On newly synthesized DNA during chromatin replication, nucleosomes are assembled from new naive histones and old modified histones. Reverón-Gómez et al revealed that H3K4me3, H3K36me3, H3K79me3, and H3K27me3 positional information is reproduced with high accuracy on newly synthesized DNA through histone recycling. Further, they found that de novo methylation to restore H3K4me3 and H3K27me3 levels occurs across the cell cycle with mark- and locus-specific kinetics. 156 Their work suggests that accurate parental histone recycling preserves positional information and allows post-translational modifications transmission to daughter cells.
Just like normal cell self-renewal, cell division is also a way for NOC to transform into TIC. In most cases, a small number of tumorigenic driver mutations cannot cause subversive changes in tumor epigenetics. Under the premise of obtaining the advantage of clonal proliferation through mutation, TIC might still maintain the innate post-translational modifications from NOC through epigenetic memory. The cellular biology translated from the innate epigenetic state of key genes and pathways in TIC can merely be a continuation of their inherent biological functions in NOC, and is manifested as the aggressive biology of TIC. Compared to the TIC transformed from the differentiated normal cell, TIC from stem cell can maintain stronger stemness. Perhaps it is this stemness that makes TIC derived from stem cells more competitive and survival advantage. In this regards, it should follow Darwinian principles.
The Cellular Origin of TIC can Determine Both Tumor Parenchyma and Stroma Heterogeneity
The heterogeneity within each individual tumor is called intratumoral heterogeneity, which starts with a single TIC and becomes greater as the disease progresses. 157 Intratumoral heterogeneity is thought to play a significant role in treatment resistance and failure. Recent few years, several mathematical models were put forward to describe intratumoral heterogeneity. These models well demonstrated the intratumoral heterogeneity caused by accumulated genetic and epigenetic mutations, and microenvironment stress.15,158 Different from previous models, we currently attempted to discuss the influence of the intrinsic stemness of TIC on parenchymal and stromal heterogeneity of a tumor, and to describe the epigenetic regularity behind tumor heterogeneity.
For the epigenetic causes of intratumoral heterogeneity, cell differentiation hierarchies and tumor microenvironment are two major concerns.
159
The former is also the core idea of the TSC model. However, ignoring the cellular origin of tumors may lead to an improper estimation of intratumoral heterogeneity in an individual. Figure 2 shows the relationship between the cellular origins of tumors and the heterogeneity of the parenchymal cells within tumors. In a given system, TICs of different cellular origins possess different capacities for differentiation or self-renewal. Tumors that originate from less-differentiated stem cell types might have a greater degree of intratumoral heterogeneity because they could have had potential to produce offspring at more hierarchies of differentiation. At the other extreme, if tumors originate from mature cells, acquisition of genetic or epigenetic mutations via self-renewal may be the only way contributing to their intratumoral heterogeneity. If intratumoral heterogeneity is driven solely by random genetic- or epigenetic-alterations, then finding rules for parenchymal heterogeneity within a tumor is challenging. The high epigenetic similarity between TICs and their NOCs strongly suggests the importance of tracing the origin of tumors more precisely in practice, since numerous studies have shown that normal lineage-specific molecules and pathways can be effective targets for tumor therapy. In addition, precancerous cells derived from less-differentiated NOCs might develop into more aggressive malignancies due to their inherent stemness. The influence of cellular origin on the heterogeneity of the parenchymal cells within a tumor. Intratumoral heterogeneity begins with a TIC and increases with the expansion and evolution of tumors. In this figure, we assume that TICs from different sources have the same genetic- and epigenetic-mutation background and evolve by obtaining two additional driver mutations (in reality, the types and quantities of them are diverse). All cells contained in a dotted box represent heterogeneous cell compositions within the tumor derived from a TIC. In a given tumor entity, TIC can originate from multiple cell types. Tumors derived from less-differentiated NOCs can have a greater degree of intratumoral heterogeneity, because they can produce offspring with the same genetic or epigenetic mutations at more hierarchies of differentiation. For TICs derived from terminally differentiated cells, acquisition of genetic- or epigenetic mutations via self-renewal may be the only way to generate heterogeneous cells. Targeting TSCs are expected to be a potential therapeutic strategy for tumor eradication, and it is a common practice to use biomarkers of normal stem cells to distinguish TSCs. The diversity of the cellular origin of a given tumor and its subtypes suggests that tracing the cellular origin of tumors and killing TICs is the basis for tumor eradication.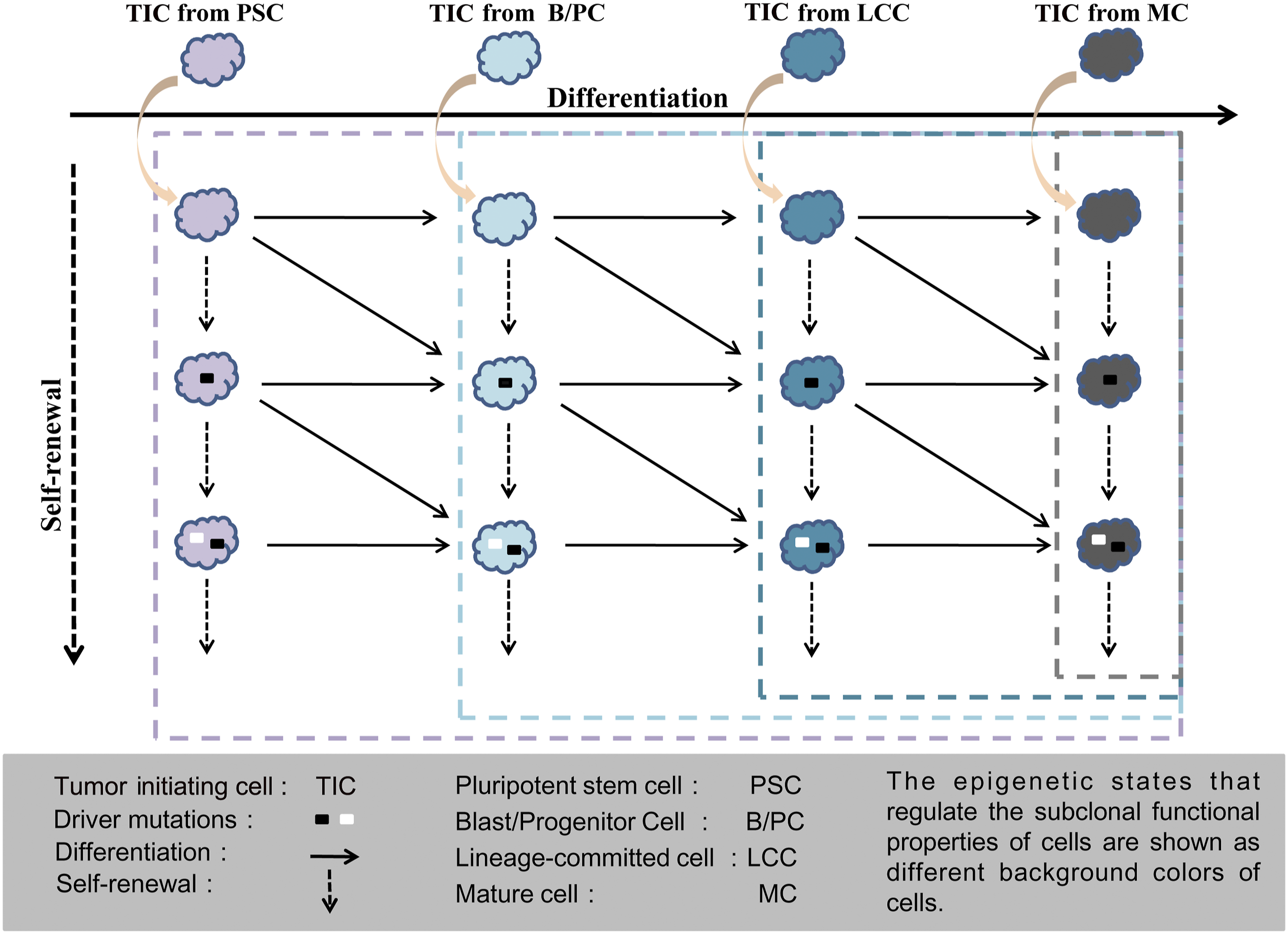
Tumor cells and their microenvironment are a complete functional system. Most of the previous studies have focused on the influence of microenvironment on the acquisition of genetic and epigenetic abnormalities of TIC and the formation of tumor biology, 160 which may be one-sided for understanding the relationship between tumor microenvironment and tumor parenchyma. Normal stem cells can recruit other cells, such as myofibroblasts/fibroblasts, mesenchymal stem/progenitor cells, endothelial cells, vascular cells, adipocytes, and immune cells. All of these cells and their secreted paracrine factors, as well as the extracellular matrix, make up the stem cell microenvironment and play significant roles in fate decision of stem cells, tissue development, and homeostasis. 161 The stem cell microenvironment is diverse and dynamic, and the information it senses and conveys reflects the difference in tissue types and anatomic sites, as well as the state of differentiation.
Many parallels have been drawn between the tumor microenvironment and physiological microenvironment, including those observed in embryonic development, organogenesis, and inflammation/wound healing. A study from Lacina revealed various biological aspects of interaction between CSCs of melanoma and cancer-associated fibroblasts with multiple parallels to interaction of normal epidermal stem cells and their niche. 162 Through fueling STAT3, MAPK, and Akt signaling, elevated expression of IL-6 by mesenchymal stem cells is associated with cancer cell proliferation, angiogenesis and metastasis in neuroblastoma, breast, colon, pancreatic, esophageal, gastric, hepatic, pancreatic, non-small cell lung, renal cancer, and multiple myeloma. 163 Tissue-resident macrophages and regulatory T cells (Tregs) are well-known regulators involved in maintaining stem cell homeostasis. Interestingly, induced macrophages and Tregs are prominent in the tumor microenvironment, where they expand in response to IL-6, VEGF, CXCL12, IL-10, and TGF-β and are capable of producing a number of immunosuppressive signals. 164 Tumor angiogenesis is essential for tumor growth and metastasis. Vascular endothelial cells from the local environment and bone marrow-derived endothelial progenitor cells are recruited into tumor niches by similar proangiogenic factors in embryonic development and organogenesis to initiate and promote angiogenesis.133-136 The extracellular matrix (ECM) is composed of a variety of proteins, polysaccharides, and proteoglycans that make up the basement membrane and the interstitium. Normal stem cells can directly or indirectly recruit ECM to maintain stemness and avoid immune clearance. In liver, abundant fibrous stroma is usually deposited in those tumors with stem cell phenotypes, such as neuroendocrine carcinoma, hepatoblastoma, scirrhous hepatocellular carcinoma, and fibrolaminar carcinoma, rather than well-differentiated hepatocellular carcinoma and adenoma. 165 As such, it cannot be ruled out that the components of a TIC’s microenvironment, if not all the components, can be proactively recruited by TICs, which may be dominated by hereditary epigenetic mechanisms. Furthermore, differences in the origin of TICs may determine the differences in tumor microenvironmental components associated with treatment or prognosis, including inflammatory cell subsets, fibroblasts, and secreted products (such as cytokines and chemokines) as well as non-cellular components of the extracellular matrix, which has important implications for understanding and evaluating the effectiveness and drug resistance of therapies that targeting tumor stroma, and will be discussed in the next section.
Conclusions
Mutations are indeed critical to tumorigenesis and are associated with clinical behavior. However, in a given tumor, not all of the established physiological processes are disrupted. Since the acquisition of the clonal proliferation advantage is the prerequisite for judging tumor transformation, whether possess molecular characteristics of normal stem cells are not indispensable for judging TICs. In spite of dramatically phenotypic changes can be observed in very few tumor cases, making their exact origin difficult to trace, in general, multi-lineage cellular origin of TIC can better explain the cellular origin of the vast majority of tumors.
Mutations can alter almost all types of physiological processes including differentiation, self-renewal, proliferation, metabolism, DNA mismatch repair, or cell-cycle transition. Differentiation arrest and reprogramming caused by mutation in tumors have been provoking scientists' great interests. Differentiation arrest was initially observed in lymphoid and hematopoietic tumors and subsequently in somatic tumors as well. The existence of TSC differentiation hierarchies in solid tumors suggests that cell-differentiation arrest hypothesis does not apply to all tumors. Broadly speaking, reprogramming can be understood as alteration of the status of certain signaling pathways and genes by genetic or epigenetic mutations. Specifically, reprogramming refer to the mechanisms by which expression of certain transcription factors, such as Yamanaka transcription factors (OSKM), renders somatic cells capability to acquire pluripotency or totipotency-like features. 166 Today, some literature suggests that cell reprogramming can play an important role in tumorigenesis. In vitro, overexpression of MYC induced epigenetic reprogramming of mammary epithelial cells into a stem cell-like state with metastatic capacity. 167 Using intestinal organoids, Heuberger showed that concomitant activation of Notch signaling and ablation of p53 induced reprogramming of specified intestinal epithelial cells into a regenerative cell state that makes them susceptible for tumorigenesis. 168 These studies on epigenetic reprogramming highlight the impact of the differentiation state on cell biological behavior and may be used to explain tumorigenesis of some tumors. However, in low-grade tumor types, such as well-differentiated HCC and thyroid tumors, multiple stem cell transcription factors and biomarkers are completely absent, indicating that cell reprogramming is not a universal mechanism for tumorigenesis in all tumor types.47,50
Traditionally, the understanding of tumor heterogeneity mostly focused on the complexity of tumor parenchymal cell composition and the disorder of signal regulation due to the introduction of genetic or epigenetic mutations or the stress of the microenvironment. Increased tumor heterogeneity of this kind is indeed strongly associated with the more aggressive biological characteristics of tumor cells, therapeutic resistance, and poor clinical outcomes for patients. However, the ultimate goal of tumor heterogeneity exploration is to find information or phenomena for clinical diagnosis and treatment. Therefore, it may be more practical to discover the underlying rules behind tumor heterogeneity.
Today, advances in large-scale genome sequencing have enabled the identification of characteristic genetic signatures in many tumors, which has become a key prerequisite for targeted drug therapy. However, genetic abnormality-targeted drugs require strict matching of mutated gene types, which can benefit only a limited group of patients. In contrast to the randomness of genetic and epigenetic mutations, the regularity of epigenetic determinants inherited from NOCs may help us to develop broad-spectrum anti-tumor drugs, as these determinants may be shared by the TICs of tumors of different systems at similar stages of differentiation. In recent years, the drugs targeting programmed cell death protein-1 (PD1)/programmed cell death protein-1 ligand 1 (PD-L1) pathway have achieved promising effects in various tumor types. PD1 and PD-L1 act as important negative immune regulators. Combining PD1 receptors on T cells with their corresponding ligands PD-L1 on normal stem cells can inhibit T cell activation and prevent the stem cells from immune killing.169,170 In some tumors, this mechanism is borrowed by tumor cells and lead to tumor cell immune escape.171,172 The purpose of PD1/PD-L1 immunotherapy is to restore the anti-tumor activity of T cells by blocking this pathway.
As in the normal tissues, the negative modulation pathway of PD1/PD-L1 exists in tumors of multiple systems, which indicates that hereditary epigenetics-based drugs have the potential to become tumor-type agnostic therapies. Even though PD-1/PD-L1 blockade have brought new hope to tumor therapy, PD-1/PD-L1 inhibitors have only benefited nearly 20% of tumor patients. Detecting the expression rates of PD-L1 (tumor cells) and PD1 (lymphocytes) in biopsy tissues is the main method to determine drug response at present, but the therapeutic effect based on this method is sometimes not ideal. 173 Although different mechanisms, such as false positive or false negative detection, irrelevant PD-1T cell expression and tumor suppression immune destruction, or lack of TNF-γ-mediated inflammation have been used to explain the limitations of this treatment, the lack of PD-L1 expression in TIC determined by its cellular origin may be one of the reasons that cannot be ignored, because in normal cells, the expression of PD-L1, as well as some tumor-associated molecules, such as histone deacetylase, DNA methyltransferases, and vascular endothelial growth factor receptors, are closely related to their differentiation stage.174-178 If TICs can express PD-L1, PD-L1 blockade may produce the best therapeutic effect. If the target molecule is only expressed in the differentiated progeny cells of TIC, the same therapy can lead to a reduction in tumor volume, but cannot eliminate TIC from the source and cause relapse. It is well established that the cytotoxic T-lymphocyte associated protein 4 (CTLA-4) pathway is another complementary immunosuppression mechanism to PD1/PDL1 pathway that allows cancer cells to escape host immunity. 143 Notably, both PD1/PDL1 and CTLA-4 pathways are critical for embryonic development, organogenesis, and physiological homeostasis in adults because they can prevent somatic cells from being destroyed by cell-mediated immune responses.139,140,179,180 Understanding the epigenetic status of these pathway in the differentiation hierarchies of normal stem cells and tracking the cellular origin of tumors more precisely may lead to increased efforts to develop combination immunotherapy strategies or to determine which population of patients can benefit from the immunotherapies. 181
Along with the effectiveness of PD-1/PD-L1 blockers, their side effects, including skin toxicity, colitis, pneumonia, liver toxicity, etc. have emerged. 182 However, it is not clear which cancer patients who use PD-1/PD-L1 blockers have potential side effects. Many tumors have a background of chronic inflammatory diseases, such as chronic hepatitis, pneumonia, and enteritis. In severe cases, stem cells can proliferate significantly to compensate for the loss of cells in the organs, and at the same time, the stem cells will recruit PD1 + lymphocytes to avoid killing themselves. Therefore, while activating the immune system to kill tumor cells, blockers in PD-1/PD-L1 pathway may also lead to the killing of normal stem cells, thus triggering or aggravating the acute inflammatory response.
There are some limitations to this work. First, based on the available literature, we only compared the similarity of partial epigenetic regulation between tumors and normal origin cells in limited systems. Systematic comparative studies of epigenetic profiles between hierarchical descendants of normal stem cells and tumors and their subtypes in different tissues are necessary. Second, although the term of epigenetic abnormality is widely used, it is not clear how many reported differential epigenetic events/epigenetic abnormalities in tumors are stochastic epimutations rather than hereditary epigenetic events from NOCs. Given the dynamic regulation of gene expression by epigenetic mechanisms, epigenomic analysis of single cells of different differentiation stages will provide more accurate evidence for understanding the global patterns of epigenetic regulation in cancers. Third, genetic and epigenetic determinants are likely to interact in creating a TIC’s hallmark. It is unclear to what extent they synergistically endow TICs with a malignant phenotype. Finally, in the in vitro system, genetic manipulation can help differentiated cells to regain stemness, which have not been proven in vivo. Whether epimutations can cause dedifferentiation in vivo is unknown.
In a given system, the cellular origin of TIC can not only determine the heterogeneity between tumors or tumor subtypes, but also determine the heterogeneity of the parenchyma and stroma within the tumor, which may be dominated by the innate epigenetic status of genes and pathways from NOC through epigenetic memory. Although tumor heterogeneity owing to the genetic or epigenetic determinants raises diagnosis and treatment challenges, tumor heterogeneity is not always a random and unpredictable phenomenon. An in-depth understanding of the global patterns of both genetic and epigenetic aspects of tumors will enable us to accurately depict the essential characteristics of tumors, which is expected to open a new path for tumor classification, prognosis assessment, individualized treatment, and new target discovery in the future.
Footnotes
Acknowledgments
We would like to thank Professor Gaosheng Huang (Department of Pathology, Fourth Military Medical University) and Chunguang Li (Department of Pathology, the 251st Hospital of the PLA). Their profound insights and spirit of exploration in medicine have always influenced us. We also thank the editors and three anonymous reviewers for their insightful and constructive comments.
Author Contributions
J.L. Feng contributed the central idea, analyzed most of the data, and wrote the initial draft of the paper. D.W. Zhao and F.D. Lv contributed to refining the ideas, carrying out additional analyses. Z.Y. Yuan contributed to drawing the figures. All authors discussed the results and revised the manuscript.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the Scientific Research Project of Beijing You-an Hospital, CCMU (No. YNKTTS20180110).
Ethical Statement
This is a review work that does not involve ethical approval and consent to participate. All authors approved the manuscript and publication.