
Abstract
Skin cutaneous melanoma (SKCM) is a type of highly invasive cancer originated from melanocytes. It is reported that aberrant alternative splicing (AS) plays an important role in the neoplasia and metastasis of many types of cancer. Therefore, we investigated whether ASEs of pre-RNA have such an influence on the prognosis of SKCM and the related mechanism of ASEs in SKCM. The RNA-seq data and ASEs data for SKCM patients were obtained from the TCGA and TCGASpliceSeq database. The univariate Cox regression revealed 1265 overall survival-related splicing events (OS-SEs). Screened by Lasso regression, 4 OS-SEs were identified and used to construct an effective prediction model (AUC: .904), whose risk score was proved to be an independent prognostic factor. Furthermore, Kruskal–Wallis test and Mann–Whitney–Wilcoxon test showed that an aberrant splicing type of aminoacyl tRNA synthetase complex-interacting multifunctional protein 2 (AIMP2) regulated by CDC-like kinase 1 (CLK1) was associated with the metastasis and stage of SKCM. Besides, the overlapped signal pathway for AIMP2 was galactose metabolism identified by the co-expression analysis. External database validation also confirmed that AIMP2, CLK1, and the galactose metabolism were associated with the metastasis and stage of SKCM patients. ChIP-seq and ATAC-seq methods further confirmed the transcription regulation of CLK1, AIMP2, and other key genes, whose cellular expression was detected by Single Cell Sequencing. In conclusion, we proposed that CLK1-regulated AIMP2-78704-ES might play a critical role in the tumorigenesis and metastasis of SKCM via galactose metabolism. Besides, we established an effective model with MTMR14-63114-ES, URI1-48867-ES, BATF2-16724-AP, and MED22-88025-AP to predict the metastasis and prognosis of SKCM patients.
Introduction
Skin cutaneous melanoma (SKCM) is a rare cancer that accounts for 1% of all malignant tumors. Genetically, as an ever-increasing and extremely invasive type of cancer, it is caused by the malignant proliferation of melanocytes.1-3 Although surgical treatment has been shown to be successful in localized melanoma, SKCM still has a high incidence of local recurrence and distant metastasis.3,4 As a difficult stage of SKCM, metastatic SKCM often has a poor response to conventional therapies. Although the discovery of BRAF driver mutations and BRAF target inhibitors has significantly improved the prognosis of patients with metastatic melanoma, a considerable number of BRAF wild-type patients with metastatic melanoma were unable to benefit from the new target treatment.5,6 Furthermore, potential immunotherapies for BRAF wild-type individuals with metastatic SKCM, such as anti-PD-1, anti-CTLA4, and interleukin-2, do not demonstrate a long-term treatment impact.7,8 As a result, there is an urgent need to investigate the etiology and metastatic mechanisms of SKCM in order to identify novel therapeutic targets for SKCM therapy.
Alternative splicing (AS), an important post-transcriptional regulation process, effectively diversifies the structures and functions of mRNAs produced from a single gene.9,10 Furthermore, splicing factors (SFs) control alternate splicing events (ASEs), forming a complex regulatory network.11,12 There are new studies indicating a link between aberrant AS and cancer incidence, development, and resistance to treatment.13,14 The amount of SF expression has also been shown to affect the splicing patterns of numerous proteins that participate in cancer-related pathways. 15 Hence, we postulated that abnormal ASEs and SFs may serve as critical prognostic indicators and new treatment targets for patients with SKCM.
Although a thorough study of ASEs and a regulatory network of ASEs and SFs has been discovered in melanoma, metastasis-associated ASEs and prognostic signaling pathways, which are equally essential to the prognosis of SKCM, have been overlooked. 16 In the present research, an integrated bioinformatics analysis of AS profiling was conducted to discover the overall survival-associated ASEs (OS-SEs) in patients with SKCM, and a prognostic model was built to predict the survival of patients with SKCM, which may be useful in therapeutic treatment. Furthermore, Pearson correlation analysis revealed metastasis-associated ASEs, as well as regulatory SFs and signaling pathways, to uncover the underlying metastasis mechanism of SKCM, which may offer prospective biomarkers and therapeutic targets for SKCM metastasis.
Material and Methods
Data Collection
RNA sequencing data and clinical information of 262 SKCM patients were obtained from the Cancer Genome Atlas (TCGA) Database (https://tcgadata.nci.nih.gov/tcga/). Meanwhile, the gene expression level of 390 alternative SFs was extracted from the 262 patients’ RNA-seq data. The percent Spliced In (PSI) value was also imputed for seven kinds of AS events to quantify AS events.
Identification of OS-SEs
The univariate Cox model was applied to identify OS-SEs, which were illustrated in the UpSet plots. In addition, the prognosis-associated ASEs and prognosis-unassociated ASEs were both integrated in the volcano plot. Meanwhile, we selected the top 20 OS-SEs of alternative promoter (AP), exon skip (ES), alternative acceptor site (AA), mutex exon (ME), alternative terminator (AT), reserved intron (RI), and alternative donor site (AD) to show in the seven bubble plots, respectively, where the size and color of bubbles signified the value of these ASEs for overall survival (OS).
Construction of Prognostic Model
Before the multivariate Cox regression, Lasso regression was applied to screen the top 20 prognostic-associated OS-SEs to avoid overfitting of the prediction model. Then, the regression coefficient of each prognostic factor screened by Lasso regression was determined by the multivariate Cox regression, represented by β value. Thus, risk score could be obtained by the following formula

The area under ROC curve was applied to test the accuracy of the model. Based on the median value, the samples were clustered into two risk subgroups medially. Then, Kaplan–Meier survival analysis was performed to show the difference between the high- and low-risk subgroups. In addition, these patients were listed in ascending order by risk score to make the risk curve, scatter plot, and heatmap. The univariate and multivariate Cox regression analyses were performed to test whether the risk score was able to predict the survival probability independently, together with age, sex, grade, stage, and TNM stage.
Construction of Splicing Correlation Network
Pearson correlation analysis was implemented to explore the possible correlation and interaction between OS-SEs and 390 SFs retrieved in the SpliceAid2 database. After excluding regulation couples with P > .001 and the absolute value of correlation coefficient <.750, the regulation network was produced by Cytoscape (3.7.1). 17 In the network plot, SF and OS-SEs were represented by arrows and ellipses, respectively. Similarly, high and low risks of OS-SEs were defined as red and purple, respectively. Positive and negative regulations were symbolized by red and green, respectively.
We also performed Kruskal–Wallis test and Mann–Whitney–Wilcoxon test to search the OS-SEs associated with metastasis and/or TNM stage, and the results were shown in the beeswarm plots. The Venn plot was also produced to show the intersections between metastasis-related OS-SEs and stage-related OS-SEs.
Signaling Pathways Enrichment Analysis
First, we used Gene Set Variation Analysis (GSVA) to identify the signaling pathways related with prognosis. The univariate Cox analysis was then used to filter the OS-related signaling pathways. The metastatic and stage-related OS-SE, as well as the prognosis-related Kyoto encyclopedia of Genes and Genomes (KEGG) pathways, were co-expressed to discover potential downstream processes of specific OS-SE.
Online Database Validation
In order to eliminate possible bias, we also collected and analyzed the gene and protein expression levels of major biomarkers in tissues in many databases, including the human protein atlas, 18 GEPIA, 19 UCSC xena, 20 UALCAN, 21 cBioportal, 22 Oncomine, 23 CCLE, 24 STRING, and Pathcard.
Assay for Transposase-Accessible Chromatin using Sequencing (ATAC-seq) and Chromatin Immunoprecipitation sequencing (ChIP-seq)
In order to further validate the transcriptional regulation of CLK1, and other key genes (BATF2, MED22, MTMR14, and URI1), ATAC-seq and ChIP-seq were performed. First, the ATAC-seq data of SKCM patients were acquired from the TCGA database, which was used to detect the chromatin accessibility in the position of key genes. Furthermore, we tested the binding relationship between CLK1 and key genes directly using the ChIP-seq method, which utilized the Cistrome database.25,26
Single Cell Sequencing
We acquired the Single Cell Sequencing data of melanoma from Single Cell Expression Atlas and analyzed the expression of these key genes in cellular level, aiming at discovering potential cellular mechanism. 27
Statistics Analysis
In this study, we used the R software (www.r-project.org; version 3.6.1; Institute for Statistics and Mathematics, Vienna, Austria) across all statistical analyses (two-sided P value <.05 was pre-set as statistically significant).
Results
Identification of ASEs in SKCM
The flow chart illustrated the analysis process of this study (Figure 1). Supplementary Table S1 summarizes the baseline information of 470 patients diagnosed with SKCM. A total of 470 SKCM patient’s data have been identified from the TCGA database to analyze ASEs. Among these SKCM cases, a total of 41 446 ASEs in 9780 genes were identified, including 2350 AAs (202 genes), 2069 ADs (209 genes), 3273 APs (642 genes), 3614 ATs (1170 genes), 6160 ESs (1651 genes), 175 MEs (30 genes), and 1780 RIs (301 genes) (Figure 2A). Therefore, an individual gene was able to go through various types of splicing events. Obviously, ES was the most prominent splicing pattern. The flowchart of analysis process. The Upset plot of different types of ASEs in the SKCM patients derived from TCGA database (A). The Upset plot of seven types of ASEs which are associated with overall survival of SKCM (B). SKCM: Skin cutaneous melanoma; TCGA: the Cancer Genome Atlas; ASEs: alternative splicing events.

Identification of OS-SEs
According to results of the univariate Cox regression analysis, a total of 1265 ASEs were significantly related to OS, which was integrally illustrated in the Upset plot (Figure 2B). Meanwhile, the volcano plot revealed that most of the ASEs were prognosis-related in SKCM (Figure 3A). The bubble plots reported the top 20 OS-ASEs of the seven kinds of splicing patterns (Figure 3B-H). Notably, MTMR14-63114-ES, URI1-48867-ES, BATF2-16724-AP, and MED22-88025-AP were among the most significant differentially expressed OS-SEs in patients with SKCM. The volcano plot of prognosis-related ASEs in SKCM (A). The bubble plot of the top 20 OS-ASEs in seven types of alternative splicing (C-G). ASEs: alternative splicing events. SKCM: Skin cutaneous melanoma; ASEs: alternative splicing events.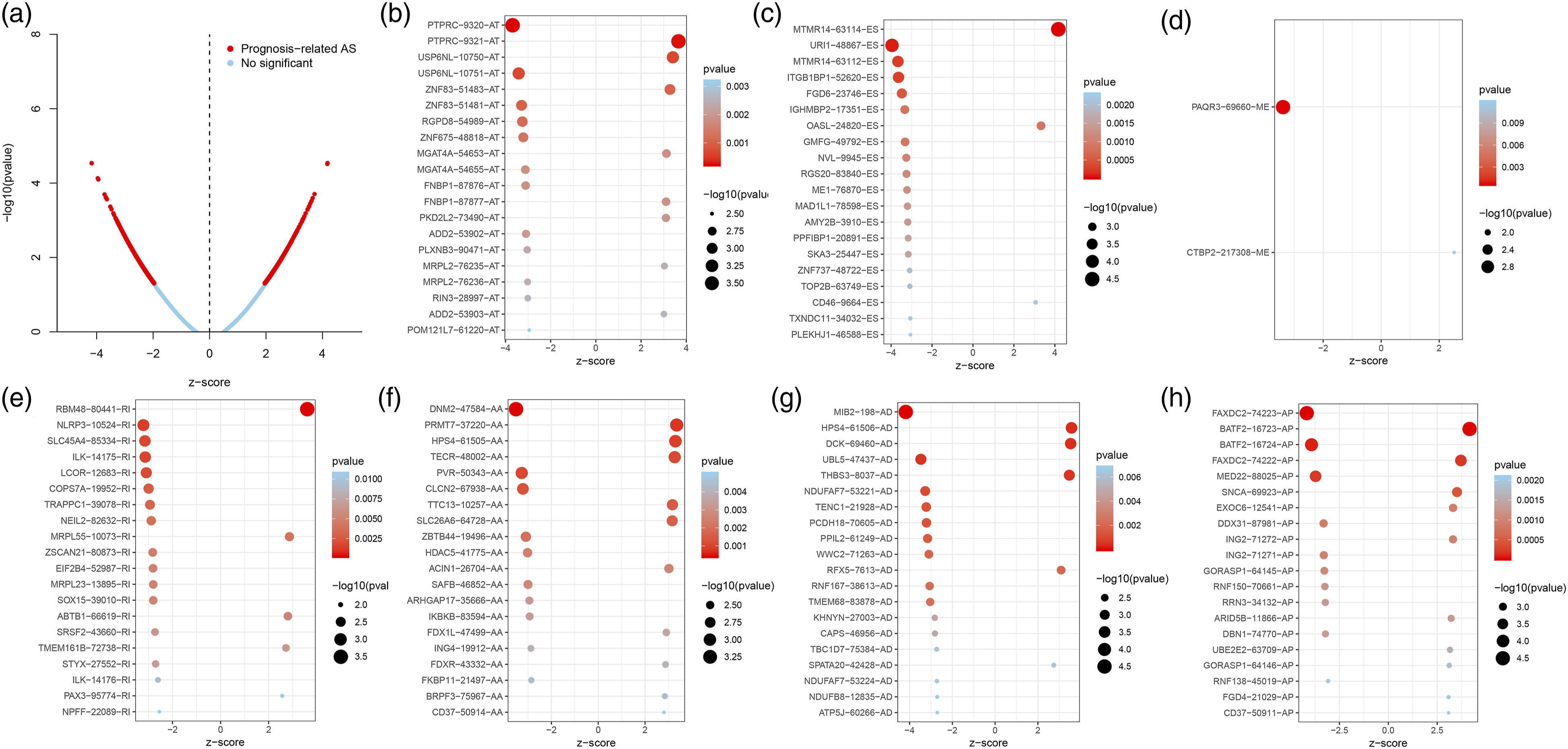
Construction of Prognostic Model for SKCM
To avoid over-fitting of the model, we implemented the Lasso regression to further screen the top 20 OS-SEs (Figures 4A and 4B). The result indicated that MTMR14-63114-ES, URI1-48867-ES, BATF2-16724-AP, and MED22-88025-AP were the most significant prognostic biomarkers. Based on the four biomarkers, we performed the multivariate Cox regression analysis to identify the value of the individual OS-SEs on the OS in patients with SKCM and constructed the corresponding predict model, whose accuracy and reliability were proved by ROC curve (AUC:0.788) (Figure 4C). Then, the risk score of each sample was obtained according to the prognosis model, with a median level of 7.168213949. The Kaplan–Meier plot indicated that there was an obvious difference in survival probability between the high- and low-risk subgroups, indicating the effectiveness of this predict model (Figure 4D). Besides, the risk curve and scatter plot showed that patients with a higher risk score tended to live longer, which also supported the validity of the model (Figures 4E and 4F). In addition, the heatmap was used to compare the expression level of OS-SEs integrated in the multivariate Cox regression. In the heatmap, MTMR14-63114-ES was lowly expressed, while URI1-48867-ES, BATF2-16724-AP, and MED22-88025-AP were highly expressed in patients with higher risk score (Figure 4G). The multivariate Cox regression model was based on ASEs selected by Lasso regression (A, B). The multivariate Cox regression model was proved to be reliable by ROC curve (AUC:0.788) (C) According to the predict model, the high-risk group in this predict model was shown to have larger survival probability (D), longer survival time (E), and mortality (F) than low-risk group. Among the four ASEs integrated in the model, MTMR14-63114-ES happened more frequently, but URI1-48867-ES, BATF2-16724-AP, and MED22-88025-AP happened less frequently in high-risk group (G).
Validation of Risk Score as an Independent Prognostic Analysis
Next, the univariate and multivariate Cox regression were applied to evaluate the independent prognostic value of risk score, together with other clinical parameters, including age, gender, grade, stage, and TNM stage. The hazard ratio in the univariate (HR = 1.060, 95%CI (1.035–1.086), P < .001) and multivariate Cox regression (HR = 1.065, 95%CI (1.029–1.103), P < .001) analyses justified that the risk score could be regarded as the independent predictor (Figure 5). In the independent prognostic factor test, the hazard ratios of risk score in the univariate and multivariate Cox regression analyses were (HR = 1.060, 95%CI(1.035–1.086), P < .001) (A) and (HR = 1.065, 95%CI(1.029–1.103), P < .001) (B), respectively.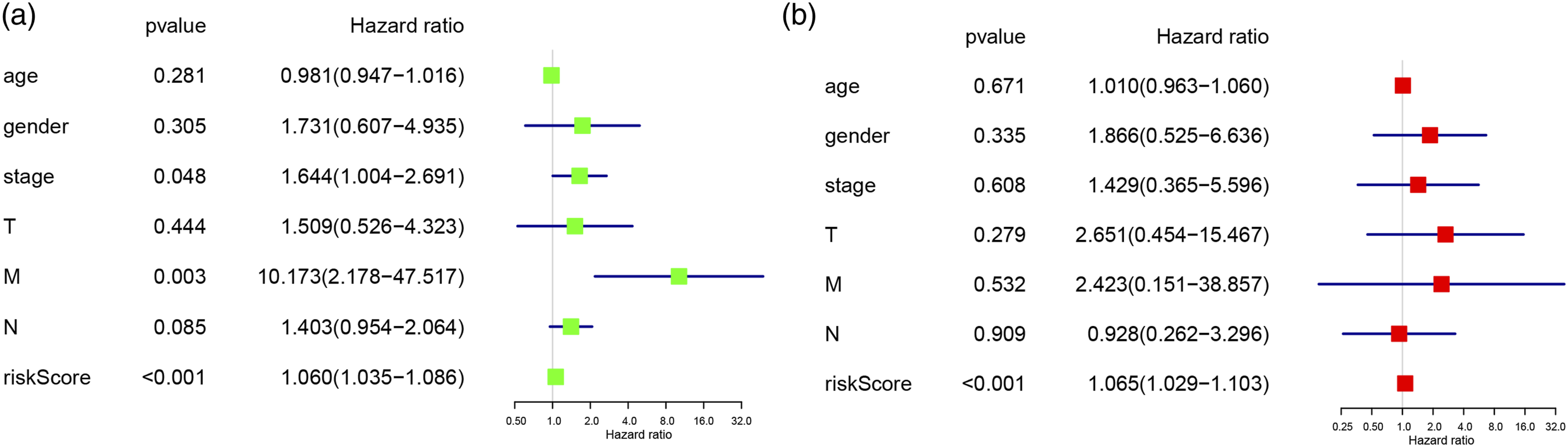
Correlation of OS-SEs and SF Expression
With regard to the regulatory SF of aminoacyl tRNA synthetase complex-interacting multifunctional protein 2 (AIMP2)-78704-ES, the network plot indicated that AIMP2-78704-ES (the high risk OS-SE, red ellipse) was regulated positively (red lines) by both CDC2-like kinase 1 (CLK1) and SRSF11 (Figure 6A). To explore the OS-SEs related to both nodal and distant metastases, we performed univariate Cox analysis and created a Venn plot to illustrate the overall results, indicating that AIMP2-78704-ES was the only OS-SEs reaching the screening standard (Figures 6B-6D). The splicing correlation network in SKCM (A) SF and OS-SEs were represented by arrows and ellipses separately. Similarly, high- and low-risk of OS-SEs were defined as red and purple and positive and negative regulations were symbolized by red and green, respectively. With the result of Kruskal–Wallis test and Mann–Whitney–Wilcoxon test (C, D), AIMP2-78704-ES was identified as the only ASE associated with both metastasis and TNM stage in the Venn plot (B). SKCM: Skin cutaneous melanoma; ASEs: alternative splicing events.
Comprehensive Analysis of ASEs and Signaling Pathways
The results of the GSVA and the univariate Cox regression analysis showed that a total of 185 KEGG pathways were related to OS. Then, the Pearson correlation analysis between AIMP2-78704-ES and all the OS-related KEGG pathways was performed to search for their co-expression association. The results revealed that AIMP2-78704-ES was significantly correlated with the galactose metabolism pathway (R = −.41, P < .001) (Figure 7). Galactose metabolism pathways were found to be correlated with AIMP2-78704-ES.
External Validation
To decrease bias induced by limited samples and vacant experimentally mechanism evidence, we used multiple online database to strength the reliability of our bioinformatics analysis. First, protein–protein interaction (PPI) network retrieved from Pathcard database suggested that Glucose-6-Phosphate Isomerase (GPI), Hexokinase 3 (HK3), Glycogen Synthase 1 (GYS1), Glucose-6-Phosphatase Catalytic Subunit 3 (G6PC3), and Beta-1,4-Galactosyltransferase 2 (B4GALT2) were the key members in galactose metabolism pathway (Supplementary Figure S1A). Besides, STRING database showed that CLK1 and AIMP2 were closed connected via galactose metabolism pathway (Supplementary Figure S1B). Then, Oncomine database illustrated that AIMP2, GPI, HK, GYS1, and G6PC3 were differentially expressed between SKCM and normal tissues (Supplementary Figure S2). CCLE and the human protein atlas provided the evidence from the aspect of cell lines and protein (Supplementary Figures S3 and S4). UALCAN database showed that CLK1, HK3, and G6PC3 were differentially expressed between localized and metastatic tumor (Supplementary Figures S5A-S5C), and that AIMP2, GPI, HK3, GYS1, G6PC3, and B4GALT2 were significantly related to OS (Supplementary Figures S5D-S5I). The expression heatmap provided by UCSC Xena uncovered the relationship between the expression of key biomarkers and OS (Supplementary Figure S6A). Besides, AIMP2, GPI, HK3, and B4GALT2 were found significantly related to patients’ OS (Supplementary Figure S6). Validation from GEPIA suggested that CLK1, GPI, HK3, and G6PC3 were differentially expressed between normal and cancer cells (Supplementary Figure S7). Ultimately, both GEPIA and cBioportal illustrated the close correlation between AIMP and other key biomarkers, and the relationship between gene expression and patients’ OS (Supplementary Figures S7 and S8). Supplementary Table S2 summarizes of multidimensional external validation results base on multiple databases
ATAC-Seq and ChIP-Seq Verification
ATAC-seq and ChIP-seq methods were utilized to verify the transcription regulation between CLK1 and key genes in the study. The results of ATAC-seq indicated that the chromatin regions in the locations of CLK1, AIMP2, BATF2, MED22, MTMR14, and URI1 were open and accessible (Supplementary Figure S9). In addition, the ChIP-seq data retrieved from the Cistrome database further revealed the DNA fragments binding with CLK1 in mouse model. The results reported strong binding peaks in the location of AIMP2, BATF2, MED22, MTMR14, and URI1 (Supplementary Figure S10). Overall, ATAC-seq and ChIP-seq together validated the binding relationship of CLK1 and other key genes in chromosomal level.
Single Cell Sequencing Verification
In order to discover the cellular expression of these key biomarkers, we analyzed the single cell sequencing data of melanoma obtained from Single Cell Expression Atlas and detected the expression of AIMP2, CLK1, BATF2, MED22, MTMR14, and URI1 (Supplementary Figures S11A and S11B) in cellular level. The results revealed that AIMP2 was highly expressed in cancer-associated fibroblasts and tumor endothelial cells, and CLK1 was highly expressed in lymph node T cells, tumor T cells, and B cell. (Supplementary Figures S11C and S11D). The cellular expression of BATF2, MED22, MTMR14, and URI1 was also reported by Single Cell Sequencing method (Supplementary Figures S11E-S11H).
Discussion
SKCM, as a highly invasive cancer, has been increasingly escalated lately.28,29 Multiple studies revealed that the prognosis of SKCM patients was closely associated with distant metastasis, and those with three or more metastatic sites usually died within one year. 30 Although multidisciplinary therapies were proposed to improve the OS of melanoma patients, most patients with metastatic SKCM still had limited efficacy. 31 Recently, many studies revealed that AS and SFs played important roles in cancer biology and had the potentials to act as the prognostic signature for tumor progression.10,11,32 However, how AS and SFs functioned in the tumorigenesis, progression, and metastasis of SKCM was still unclear. In the present study, we found out that regulatory mechanism between AIMP2-78704-ES and its critical SF (CLK1) was actively involved in tumor metastasis and TNM stage.
CLK1, composed of 454 amino acids, can auto-phosphorylate on serine, tyrosine, and threonine residues and phosphorylate exogenous substrates on serine and threonine residues. 33 AS was regulated by SFs whose activity was in turn regulated by phosphatases and splice factor kinases.34,35 Without exception, CLK1 phosphorylation of serine/arginine-rich proteins was also proved to be central to RNA splicing reactions, and actively got involved in a myriad of normal physiology and diseases, including cancer.36,37 Yuying Liu et al 38 found that phosphorylation regulation of CLK1 on eight serine residues of alternative splicing factor 45 (SPF45) was positively correlated with enhanced cell migration and invasion capability of ovarian cancer cells. In addition, the expression of CLK1 could also be induced by hypoxia in prostate cancer cells PC3. 39 In our study, we discovered that CLK1 could regulate the pre-mRNA splicing of AIMP2 to promote metastasis in patients with SKCM, which was in high accordance with the previous studies. Therefore, the inhibition of CLK1 may become a novel therapeutic target for cancer by selectively reducing some cancer-relevant proteins. 40
AIMP2 was a cytoplasmic protein acting as a non-enzymatic scaffold factor of the multi-tRNA synthetase complex (MSC), which was required for assembly and stability of the complex.41,42 Previous studies showed that AIMP2 might serve as an important molecule in regulating cell proliferation and apoptosis after DNA damage through interacting with p53.41,43 Moreover, AIMP2-DX2, one of variant splicing isoform of AIMP, was significantly associated with the tumorigenesis, cancer cell proliferation, invasion, and migration in many cancers, including lung cancer, ovarian cancer, and nasopharyngeal carcinoma44-47 In the study of Yin K et al, elevating the expression of AIMP2 splicing variation could shorten the survival of patients with tongue squamous cell carcinoma, which was also in high accordance with our study. 48 Taken together, AIMP2 may also serve as a novel therapeutic target of SKCM.
To further investigate the mechanisms of CLK1 in regulating AIMP2-78704-ES, we performed the Pearson correlation analysis between OS-related KEGG signaling pathways and AIMP2-78704-ES, and found out that abnormal ASE of AIMP2 might influence patients’ prognosis via galactose metabolism pathway. Galactose was a natural aldohexose usually presenting in the form of D-configuration. It was known that D-galactose was a common substance in bacteria, plants, and animals, and that galactose was metabolized through the Leloir pathway, which required a close cooperation of multiple metabolic enzymes. 49 The mis-regulation or malfunction of any component of metabolism pathway could result in the accumulation of toxic intermediate products and damage to cells. 50 Through PPI network of galactose metabolism retrieved from Pathcard database, we found out that Glucose-6-Phosphate Isomerase (GPI), Hexokinase 3 (HK3), Glycogen Synthase 1 (GYS1), Glucose-6-Phosphatase Catalytic Subunit 3 (G6PC3), and Beta-1,4-Galactosyltransferase 2 (B4GALT2) were the key members in the galactose metabolism pathway. A myriad of studies have uncovered the function of these 5 key biomarkers in tumorigenesis, progression, invasion, and migration.51,52 Moreover, through multiple dimensions validation from different online databases, we also found out that GPI, HKS, GYS1, G6PC3, and B4GALT2 were significantly related to OS in patients with SKCM, and these biomarkers were also greatly correlated with AIMP2, indicating the reliable analysis of our study.53,54
Despite the thorough bioinformatics analysis, our research has several limitations. First, the sample information and sequencing data were mainly acquired from Western authorities, leaving adequate information on Asian individuals unfilled. Second, although we utilized several databases to identify gene and protein expression levels of important biomarkers at the tissue and cellular levels to reduce bias (Supplementary Figures S1-S8), this was a correlation research from many dimensions rather than a biological mechanism study with exact experiment. Despite its limitations, this research did build an efficient model to predict SKCM patient survival based on four major OS-SEs and concluded that the mechanism of CLK1 in regulating AIMP2-78704-ES may play an essential role in SKCM metastasis. Importantly, in order to further investigate the relevant molecular process and validate our theory, we will conduct rigorous cell, animal, and clinical studies in the future.
Conclusion
We established an effective model with MTMR14-63114-ES, URI1-48867-ES, BATF2-16724-AP, and MED22-88025-AP to predict the metastasis and prognosis of SKCM patients. Through the bioinformatics analysis, we discovered that CLK1 might regulate AIMP2-78704-ES via galactose metabolism pathway in tumorigenesis, metastasis, and poor clinical outcomes of patients with SKCM.
Supplemental Material
sj-pdf-1-ccx-10.1177_10732748211051554 – Supplemental Material for The Identification of Prognostic and Metastatic Alternative Splicing in Skin Cutaneous Melanoma
Supplemental Material, sj-pdf-1-ccx-10.1177_10732748211051554 for The Identification of Prognostic and Metastatic Alternative Splicing in Skin Cutaneous Melanoma by Runzhi Huang, Mingxiao Li, Zhiwei Zeng, Jie Zhang, Dianwen Song. Peng Hu, Penghui Yan, Shuyuan Xian, Xiaolong Zhu, Zhengyan Chang, Jiayao Zhang, Juanru Guo, Huabin Yin, Tong Meng and Zongqiang Huang in Cancer Control
Footnotes
Acknowledgments
We thank the Cancer Genome Atlas (TCGA) team for using their data.
Author Contributions
Conception/design: Runzhi Huang, Mingxiao Li, Zhiwei Zeng, Jie Zhang, Dianwen Song, Peng Hu, Penghui Yan, Shuyuan Xian, Xiaolong Zhu, Zhengyan Chang, Jiayao Zhang, Juanru Guo, Huabin Yin, Tong Meng, and Zongqiang HuangCollection and/or assembly of data: Runzhi Huang, Mingxiao Li, Zhiwei Zeng, Jie Zhang, Dianwen Song, Peng Hu, Penghui Yan, Shuyuan Xian, Xiaolong Zhu, Zhengyan Chang, Jiayao Zhang, and Juanru GuoData analysis and interpretation: Runzhi Huang, Mingxiao Li, Zhiwei Zeng, Jie Zhang, Dianwen Song, Peng Hu, Penghui Yan, Shuyuan Xian, Xiaolong Zhu, Zhengyan Chang, Jiayao Zhang, Juanru Guo, Huabin Yin, Tong Meng, and Zongqiang Huang Manuscript writing: Runzhi Huang, Mingxiao Li, Zhiwei Zeng, Jie Zhang, Dianwen Song, Peng Hu, Penghui Yan, Shuyuan Xian, Xiaolong Zhu, Zhengyan Chang, Jiayao Zhang, Juanru Guo, Huabin Yin, Tong Meng, and Zongqiang Huang Final approval of manuscript: Runzhi Huang, Mingxiao Li, Zhiwei Zeng, Jie Zhang, Dianwen Song, Peng Hu, Penghui Yan, Shuyuan Xian, Xiaolong Zhu, Zhengyan Chang, Jiayao Zhang, Juanru Guo, Huabin Yin, Tong Meng, and Zongqiang Huang
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was supported in part by the National Natural Science Foundation of China (Grant No. 81702659; 82173168, 81772856, 82073207; 81801620;); Youth Fund of Shanghai Municipal Health Planning Commission (No.2017YQ054); Shanghai Municipal Health Commission (Grant No. 201940306); Shanghai Rising-Star Program (No. 21QA1407500); Henan medical science and technology research project (Grant No. 201602031).
Ethics Approval
The study was approved by the Ethics Committee of the First Affiliated Hospital of Zhengzhou University (approval no. 2020-KY-190a).
Informed Consent
All patients provided written informed consent prior to enrollment in the study.
Data Availability
Supplementary Material
Supplementary material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.