
Abstract
Background
AbobotulinumtoxinA (aboBoNT-A) is approved worldwide in indications related to excessive muscle activity, including focal upper and lower limb spasticity.
Objective
We evaluated real-world outcomes associated with on-label use of aboBoNT-A for adult focal spasticity in UK routine clinical practice.
Methods
This was a multicenter, retrospective, 52-week study analyzing data from patients who had received ≥1 injection of aboBoNT-A in line with the Summary of Product Characteristics. Primary endpoints were the average aboBoNT-A dose received per treatment cycle and average interval between cycles.
Results
For the 108 patients included, the median (Q1, Q3) age was 52.5 (41.0, 69.0) years and the most common underlying neurological condition was stroke (61.1%). The mean (SD) total dose of aboBoNT-A received was 540.8 (268.5) U (cycle 1), 586.1 (279.4) U (cycle 2), 558.0 (258.5) U (cycle 3), and 464.3 (209.6) U (cycle 4). The mean (SD) interval between cycles was 153.8 (56.9) days. Of 82 goals assessed, 71 (86.6%) were achieved. Seven patients (5.8%) in the safety population (N = 121) experienced adverse events, none of which were considered related to aboBoNT-A treatment.
Conclusions
These data further document aboBoNT-A as an effective and well-tolerated treatment for adult focal spasticity, supporting use in clinical practice.
Keywords
Introduction
Spasticity is a sensorimotor disorder characterized by the intermittent or sustained involuntary contraction of muscles (Pandyan et al., 2005). Symptoms of spasticity include reduced range of movement, muscle spasms, and pain (Keam et al., 2011). Furthermore, secondary complications arising from spasticity (e.g., deformity, pain, and impaired function) may interfere with rehabilitation (Royal College of Physicians, 2018). Spasticity affects an estimated 42% of post-stroke patients and 19% of those with severe traumatic head injury (Verplancke et al., 2005; Wissel et al., 2013). Therefore, effective treatment can help to minimize the impact on patient quality of life and reduce the cost of long-term care (Baricich et al., 2023; Danchenko et al., 2021).
Limb spasticity is primarily managed with physical therapy, but this can be supplemented with pharmacological interventions if necessary (Ward, 2002). Although oral antispasmodic drugs can reduce spasticity, they may cause generalized muscle weakness that can lead to further loss of function; therefore, botulinum neurotoxins are regarded as the treatment of choice for focal, multi-focal, and segmental disabling spasticity in most guidelines and consensus statements (Biering-Soerensen et al., 2022; Royal College of Physicians, 2018; Turner-Stokes & Ward, 2002). AbobotulinumtoxinA (aboBoNT-A) is globally approved for treating excessive muscle activity, including focal upper limb spasticity (ULS) and lower limb spasticity (LLS) (Electronic Medicines Compendium, 2023a, 2023b; U.S. Food and Drug Administration, 2020). When injected intramuscularly, aboBoNT-A blocks acetylcholine release at the neuromuscular junction, to generate a focal weakening of voluntary and involuntary muscle contraction (Keam et al., 2011; Royal College of Physicians, 2018). The optimal aboBoNT-A dose in patients with residual voluntary movements in the upper limb is reported to be 1000 U (Bakheit et al., 2000). Relief of symptoms typically occurs 2–5 days after injection and can last for 3–4 months, after which repeat injections can be administered to maintain response (Electronic Medicines Compendium, 2023a, 2023b; Keam et al., 2011).
Despite aboBoNT-A having been available since 1990, real-world data from large and long-term retrospective databases concerning its use as a treatment for adult focal spasticity are scarce, particularly in the UK. Local real-world data are playing an increasingly important role in supporting clinical practice and influencing commissioning considerations. Furthermore, it is important to know whether real-world prescribing aligns with the licensed indication. This is particularly relevant in the UK as it is one of the few countries where most patients receive drugs for free on the National Health Service, whereas in other countries off-label prescribing may not occur if insurance reimbursement is jeopardized. Therefore, the purpose of this study was to retrospectively analyze the real-world clinical experience associated with aboBoNT-A treatment within its licensed indication, for adult ULS and/or LLS across multiple National Health Service centers, with a focus on the outcomes and dosing information collected for initial and repeated treatment.
Materials and Methods
Study Design and Patients
This was a retrospective, longitudinal chart review study (NCT04604379) conducted at three UK National Health Service hospitals: Colman Hospital, King's College Hospital, and York Hospital. All collected data were originally recorded prior to, and independently of, the study. The prescription and administration of aboBoNT-A were carried out according to the routine clinical practice at the participating centers. Although injection sites could be determined by palpation, the licensed indication recommended the use of an injection guiding technique (e.g., electromyography, electrical stimulation, or ultrasound) (Electronic Medicines Compendium, 2023a, 2023b; U.S. Food and Drug Administration, 2020). Relevant data collected as part of the patient's routine medical care were entered into an electronic case report form (eCRF) designed specifically for the purpose of the study. eCRFs were pseudo-anonymized (given a unique patient study number), transmitted to external analysts, and stored in the same database for data analysis.
Adult patients (≥18 years old at the time of their first injection) diagnosed with focal ULS and/or LLS were eligible for this study if they were prescribed aboBoNT-A in line with the Summary of Product Characteristics (SmPC) and initiated treatment for focal ULS after January 31, 2016, and/or for focal LLS after December 6, 2016. Data cut-off was December 31, 2019. Patients had to be naïve to treatment with any type of botulinumtoxinA during the 6 months before initiation of aboBoNT-A and had to have received ≥1 treatment cycle of aboBoNT-A during the observation period, according to the SmPC (in line with recommended indications, muscles injected, dosing, and with an interval of ≥12 weeks between injections) (Electronic Medicines Compendium, 2023a, 2023b). Additionally, patients were required to be treated at the participating center for the duration of the observation period and have data recorded in their medical records available for review. Several parameters were routinely collected, but it was expected that each center would prioritize different parameters due to local practices. Patients were excluded from the study if they were participating or had participated in an interventional clinical trial of an investigational medicinal product for spasticity, which may have confounded the collection of real-world data.
The study included all specified data from eligible patients from the first injection of aboBoNT-A for focal ULS and/or LLS (the index date) for a minimum of one treatment cycle and a maximum of 52 (+6) weeks from first injection (Figure 1). The baseline period was defined as the 6-week period prior to and including the index date to allow for assessments before first injection. If multiple measurements for a single datapoint were taken during the baseline period, only the measurement on or closest to the index date was recorded. A treatment cycle was defined as the visit for aboBoNT-A administration and the follow-up visit to assess effectiveness. It was anticipated that up to four treatment cycles may occur within the observation period, consistent with the aboBoNT-A SmPC, which recommends an interval between injections of ≥12 weeks (Electronic Medicines Compendium, 2023a, 2023b).
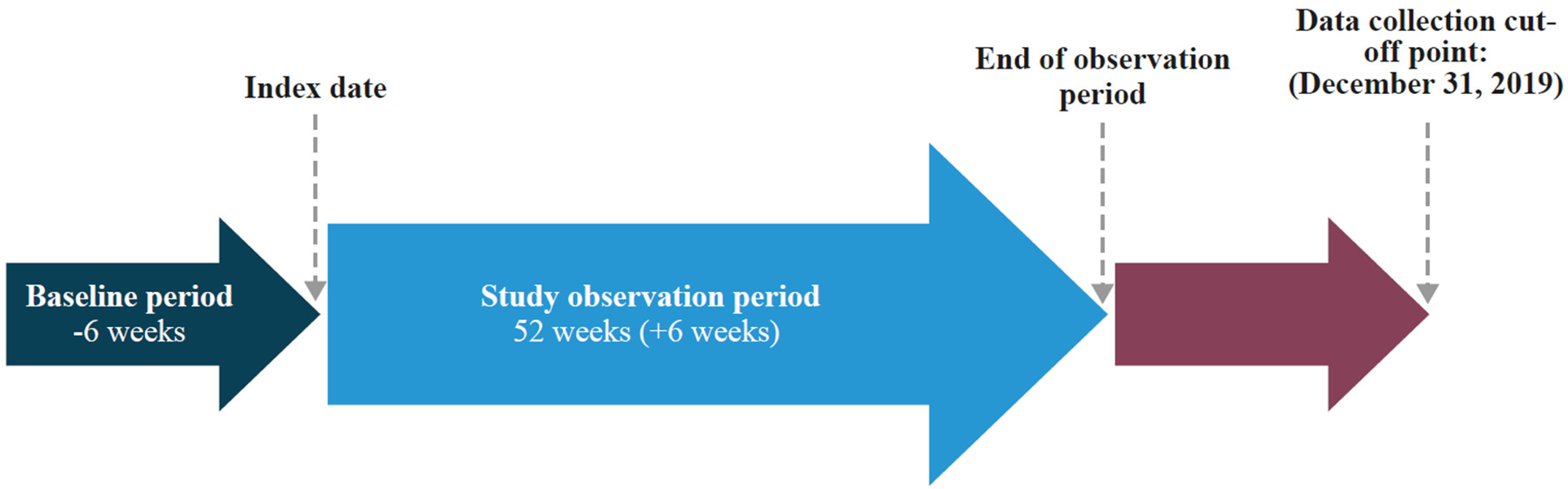
Study design. The index date was defined as the timepoint at which each patient received their first aboBoNT-A injection (after January 31, 2016, for focal ULS or December 6, 2016, for focal LLS). aboBoNT-A, abobotulinumtoxinA; LLS, lower limb spasticity; ULS, upper limb spasticity.
Outcomes
The primary objective of the study was to describe the dose and injection interval of aboBoNT-A with single and repeated injection cycles for the treatment of adults with focal ULS and/or LLS. Secondary objectives included analyses of demographic and clinical characteristics at baseline, additional injection parameters, clinical outcomes, and safety. The following clinical outcome measures were recorded if assessments were undertaken during routine practice: Goal Attainment Scaling (GAS) (Turner-Stokes et al., 2010), Modified Ashworth Scale (MAS) (Bohannon & Smith, 1987), active range of motion (AROM), Tardieu Scale, and pain visual analog scale (VAS). Safety assessments were based on data recorded in the eCRF for the observation period.
The outcomes for the safety analysis were adverse events (AEs) and special situations. All AEs recorded in the eCRF were coded using Medical Dictionary for Regulatory Activities coding, version 24.0 March 2021. In addition to overdose collected in the special situation form in the eCRF, treatment cycles with total dose (U) > 1500 for LLS, > 1000 for ULS, and >1500 for ULS + LLS were considered as overdose.
Statistical Analysis
Based on previously reported values (Jost et al., 2014), for an estimated mean (standard deviation [SD]) dose of 728 (400) U of aboBoNT-A, a sample size of 100 patients would provide a confidence interval (CI) width of 160 U (CI = 648.63, 807.37), whereas a sample size of 200 patients would provide a two-sided 95% CI width of 112 U (CI = 672.22, 783.78). Therefore, the target number of patients to include in the database was 100–200; in total, data were collected from 121 eligible patients.
All treatment practice (Supplementary Table S1) and effectiveness analyses were performed on the study population, defined as patients in the database who received ≥1 injection of aboBoNT-A and met all eligibility criteria (N = 108). All safety analyses were performed on patients in the database who received ≥1 injection of aboBoNT-A (N = 121).
Data from all participating centers were pooled for analysis and endpoints were described using summary statistics for quantitative or qualitative parameters, as appropriate. Selected endpoints were also analyzed separately by location of injection. The following summary statistics were presented: n, number of patients with available value, number of missing values, arithmetic mean, SD, median, quartiles (first quartile [Q1] and third quartile [Q3]), range (minimum, maximum), 95% CI of the arithmetic mean, and 95% CI of the median. For categorical or discrete variables, the absolute and relative (percentage) numbers were presented, including 95% CI.
The analysis of secondary endpoints was performed on the study population unless otherwise stated. Summary statistics or frequency counts for baseline characteristics, including demographic data, disease history, and comorbidities were presented. The distribution of ongoing concomitant treatment for spasticity and pain (including anti-spasticity medication, pain medication and opioids, physiotherapy, occupational therapy, and surgery) was described per injection cycle using frequency and percentage of patients. The number of injection cycles per patient was described using summary statistics for both qualitative and quantitative parameters.
AEs were tabulated overall (number and percentage of patients; number of events) and classified by primary System Organ Class and Preferred Term. Frequency distributions of special situations associated with an AE and special situations not associated with an AE were tabulated.
Missing or partial data were not imputed except for the following timepoints: date of diagnosis of neurological condition, date of diagnosis of spasticity, start date of AE, and end date of AE. If data were missing for any other endpoints, the number and percentage of missing values were tabulated and reported as part of the table for each endpoint but not included in the denominator as part of percentage calculations. For dates that were ambiguous due to missing day and/or month, standard imputation was applied to derive time and duration parameters. If a patient had a significant number of missing values or questionable data, a decision was made with the sponsor regarding the handling of these data in summaries prior to the database lock. For this study, no sensitivity analyses were conducted.
Results
Baseline Characteristics
Data were collected for 121 patients, of whom 108 were included in the study population (Figure 2). The median (Q1, Q3) age at first injection was 52.5 (41.0, 69.0) years and the most common underlying neurological condition was stroke (61.1%), followed by traumatic brain injury (9.3%) (Table 1). There was a median (Q1, Q3) interval of 43.7 (11.4, 215.9) months between diagnosis of underlying neurological condition and first injection, whereas the median (Q1, Q3) interval between diagnosis of spasticity and first injection was 6.9 (3.1, 72.0) months. Overall, 65.7% of patients had ULS only, 9.3% had LLS only, and the remaining 25.0% had ULS and LLS (Table 1). Unilateral spasticity was more common than bilateral spasticity, occurring in 88.8% of patients with ULS and 75.7% of patients with LLS (Table 1).
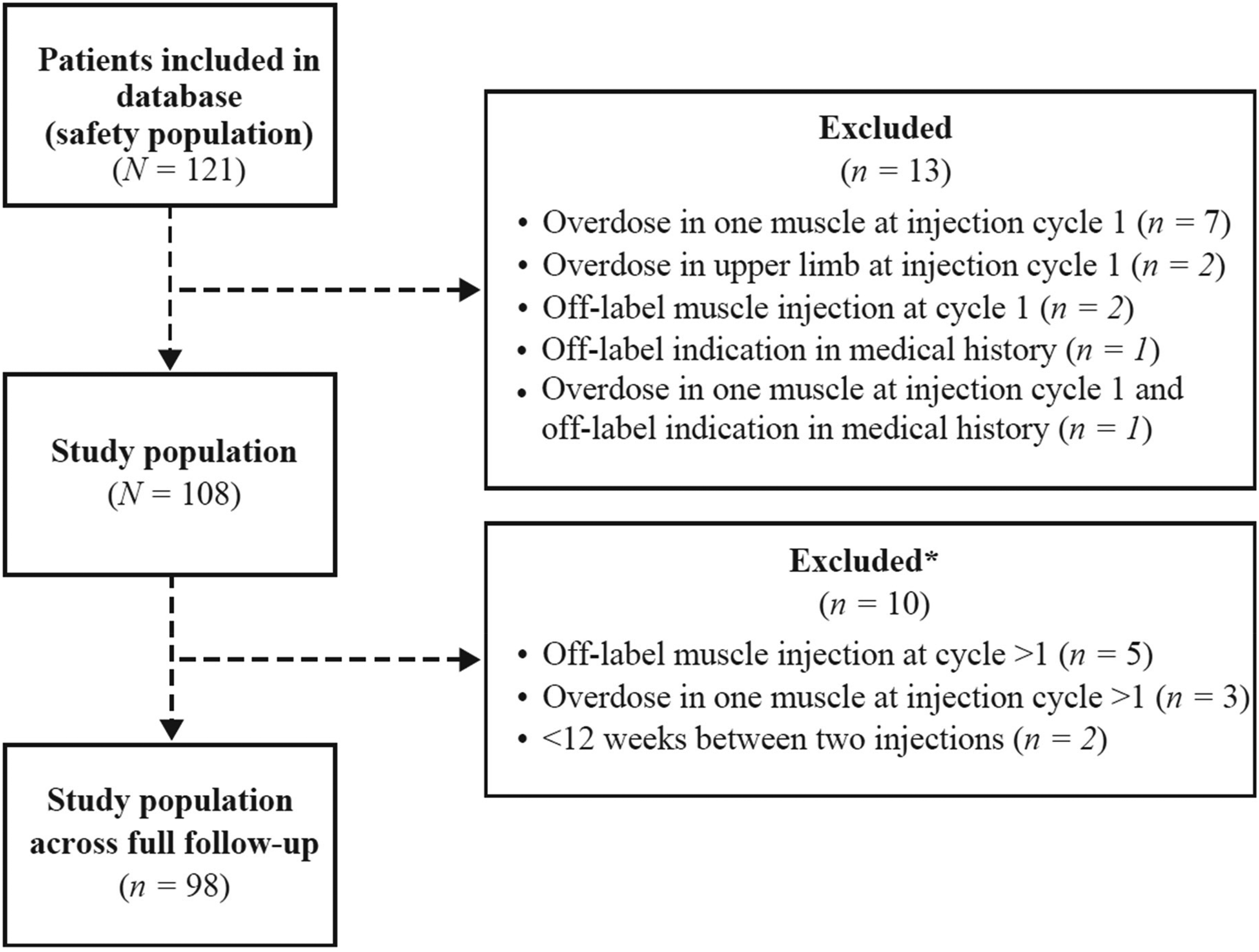
Patient disposition. *For these 10 patients, data collected up to the time of deviation from the Summary of Product Characteristics were included in the statistical analysis.
Demographic and Clinical Characteristics of the Study Population at Baseline.
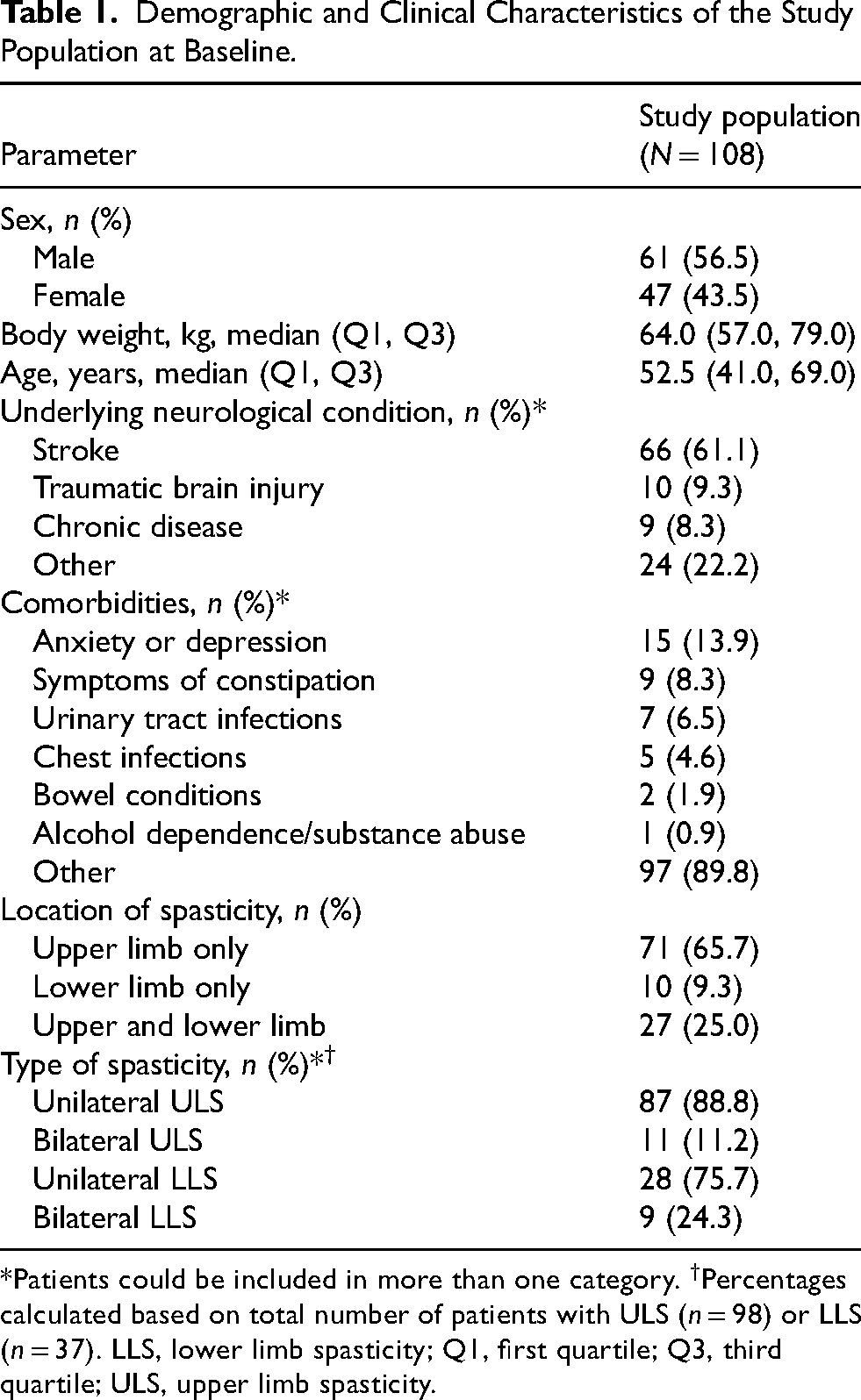
*Patients could be included in more than one category. †Percentages calculated based on total number of patients with ULS (n = 98) or LLS (n = 37). LLS, lower limb spasticity; Q1, first quartile; Q3, third quartile; ULS, upper limb spasticity.
Most patients (86.1%) had at least one ongoing concomitant treatment, including anti-spasticity medication (40.7%), pain medication or opioids (12.0%), or neurolytic agents (4.6%), or had undergone surgery (0.9%). At the time of the first injection, 29.6% and 70.4% of patients were undergoing occupational therapy and physiotherapy, respectively. Of those who received occupational therapy or physiotherapy, patients had a median (Q1, Q3) of 1.0 (0.5, 1.0) hour a week of occupational therapy and 1.0 (1.0, 1.0) hour a week of physiotherapy.
Injection Parameters
Overall, the mean (SD; 95% CI) total dose of aboBoNT-A received was 540.8 (268.5; 489.6, 592.1) U at cycle 1 (N = 108), 586.1 (279.4; 524.7, 647.5) U at cycle 2 (n = 82), 558.0 (258.5; 479.4, 636.5) U at cycle 3 (n = 44), and 464.3 (209.6; 270.4, 658.1) U at cycle 4 (n = 7) (Figure 3). The mean (SD; 95% CI) interval between treatment cycles throughout the observation period was 153.8 (56.9; 141.8, 165.8) days. The mean (SD) interval between injections 1 and 2 was 152.6 (59.4) days, between injections 2 and 3 was 131.9 (27.2) days, and between injections 3 and 4 was 113.6 (19.8) days. For patients who received injections in upper limb muscles only (n = 79), the mean (SD) interval between treatment cycles was 147.1 (50.4) days. Most patients had either two (41.7%) or three (33.3%) treatment cycles during the observation period, whereas 17.6% of patients had only one treatment cycle and 7.4% had the maximum four treatment cycles permitted in line with the SmPC.
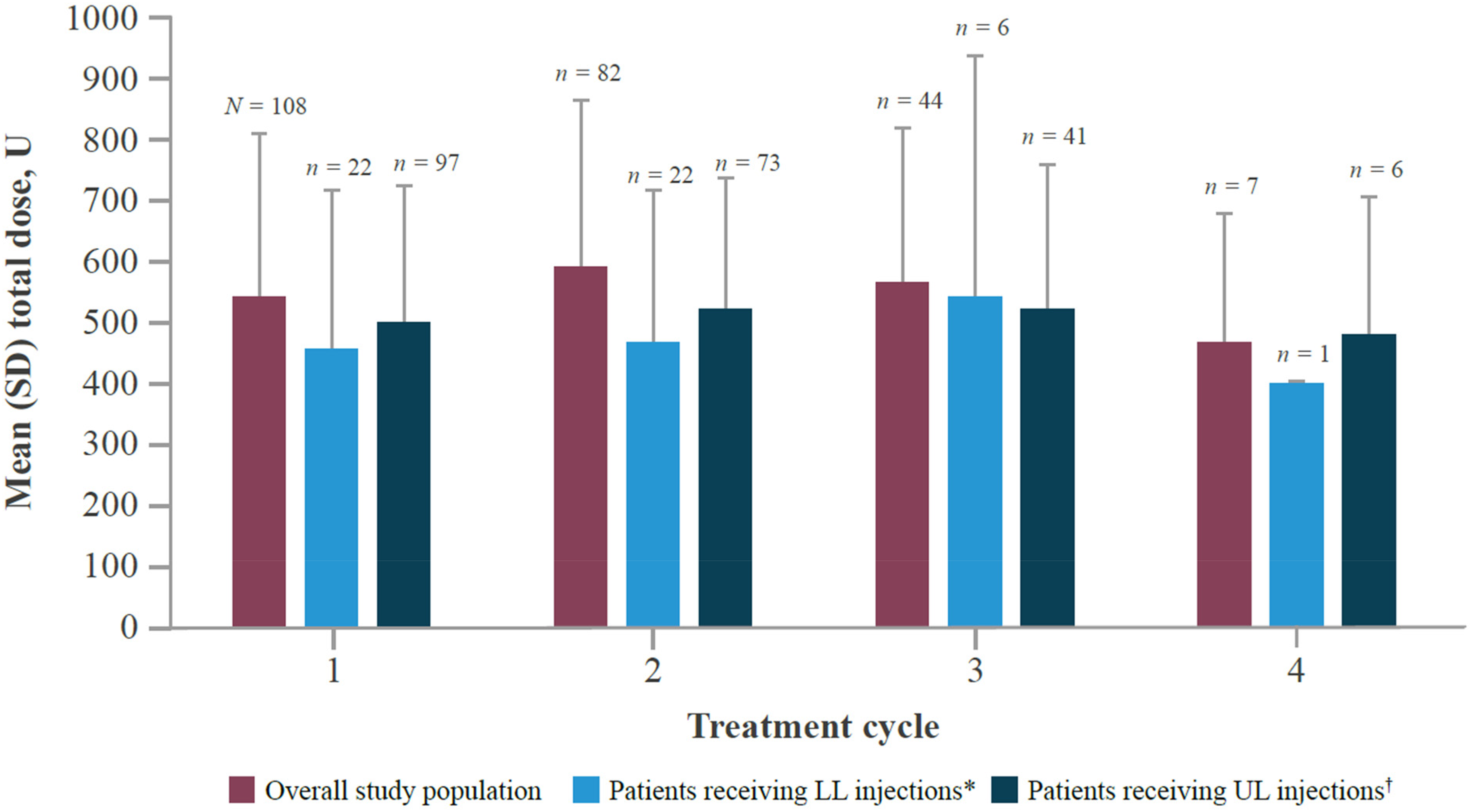
Mean total dose of aboBoNT-A received per treatment cycle. *Includes patients injected in LL only or LL + UL. †Includes patients injected in UL only or UL + LL. aboBoNT-A, abobotulinumtoxinA; LL, lower limb; SD, standard deviation; UL, upper limb.
Most patients received injections in upper limb muscles only (73.1%), followed by lower limb muscles only (9.3%) and both lower and upper limb muscles (9.3%). Additionally, 8.3% of patients received injections in lower and upper limb muscles over the observation period but not in both within the same treatment cycle. The mean (SD) number of muscles injected per treatment cycle was 3.9 (1.7). The average dose received per muscle group for each treatment cycle is presented in Table 2. For patients injected in upper limb muscles only, the mean (SD) dose ranged from 106.7 (11.5) (treatment cycle 4; n = 3; hand) to 200.0 (44.1) (treatment cycle 1; n = 19; shoulder).
Average Dose (U) per Muscle Group per Treatment Cycle.

SD, standard deviation.
The most common reasons for undergoing a second treatment cycle were recurrence of symptoms (63.6% of 22 patients who received lower limb injections; 43.8% of 73 patients who received upper limb injections) and symptom control maintenance (36.4% of 22 patients who received lower limb injections; 56.2% of 73 patients who received upper limb injections). Of the 82 patients who received ≥2 treatment cycles, 56.1% had ≥1 dose adjustment during the observation period.
Overall, 28 patients (25.9%) permanently discontinued aboBoNT-A treatment during the observation period. The reasons for discontinuation were long-lasting clinical improvement (32.1%), lack of clinical response (21.4%), loss to follow-up (14.3%), contractures (10.7%), and “other” (21.4%). The mean (SD) time to permanent discontinuation was 29.2 (12.8) weeks.
Clinical Outcomes
Set goals were recorded for 31 patients (28.7%) at the start of treatment cycle 1 and six patients (5.6%) at the start of treatment cycle 2. A set goal could be assessed multiple times throughout the observation period. Of all goals set, 59.7% related to activities or function, whereas the remaining 40.3% related to impairment or symptoms. Overall, 86.6% (71/82) of goals were achieved during the observation period. Mean (SD) GAS T-scores at the end of treatment cycles 1 (n = 29) and 2 (n = 14) were 50.7 (8.3) and 50.1 (7.8), respectively. For patients injected in upper limb muscles, the mean (SD) changes in MAS score from baseline ranged from −0.2 (0.5 and 0.7) (finger flexors [n = 41] and elbow flexors [n = 31], respectively) to 0.1 (0.2) (forearm pronators [n = 10]) at treatment cycle 2, and −0.3 (0.5) (forearm pronators [n = 8]) to −0.1 (0.4, 0.5, 0.7) (shoulder adductors [n = 7], wrist flexors [n = 25], and elbow flexors [n = 20], respectively) at treatment cycle 3 (Table 3). For patients injected in the ankle plantarflexors, the mean (SD) change in MAS score from baseline was 0.2 (0.4) at treatment cycle 2 (n = 6) and −0.3 (0.6) at treatment cycle 3 (n = 3) (Table 3). Tardieu Scale scores were recorded for a small number of patients, and of the 14 patients who had at least one primary targeted muscle group, 12 had upper limb muscles targeted, with finger flexors being the most common (n = 7). AROM and pain VAS assessments were also carried out in a minority of patients (Supplementary Tables S2 and S3).
Average MAS Score per Muscle Group per Treatment Cycle.
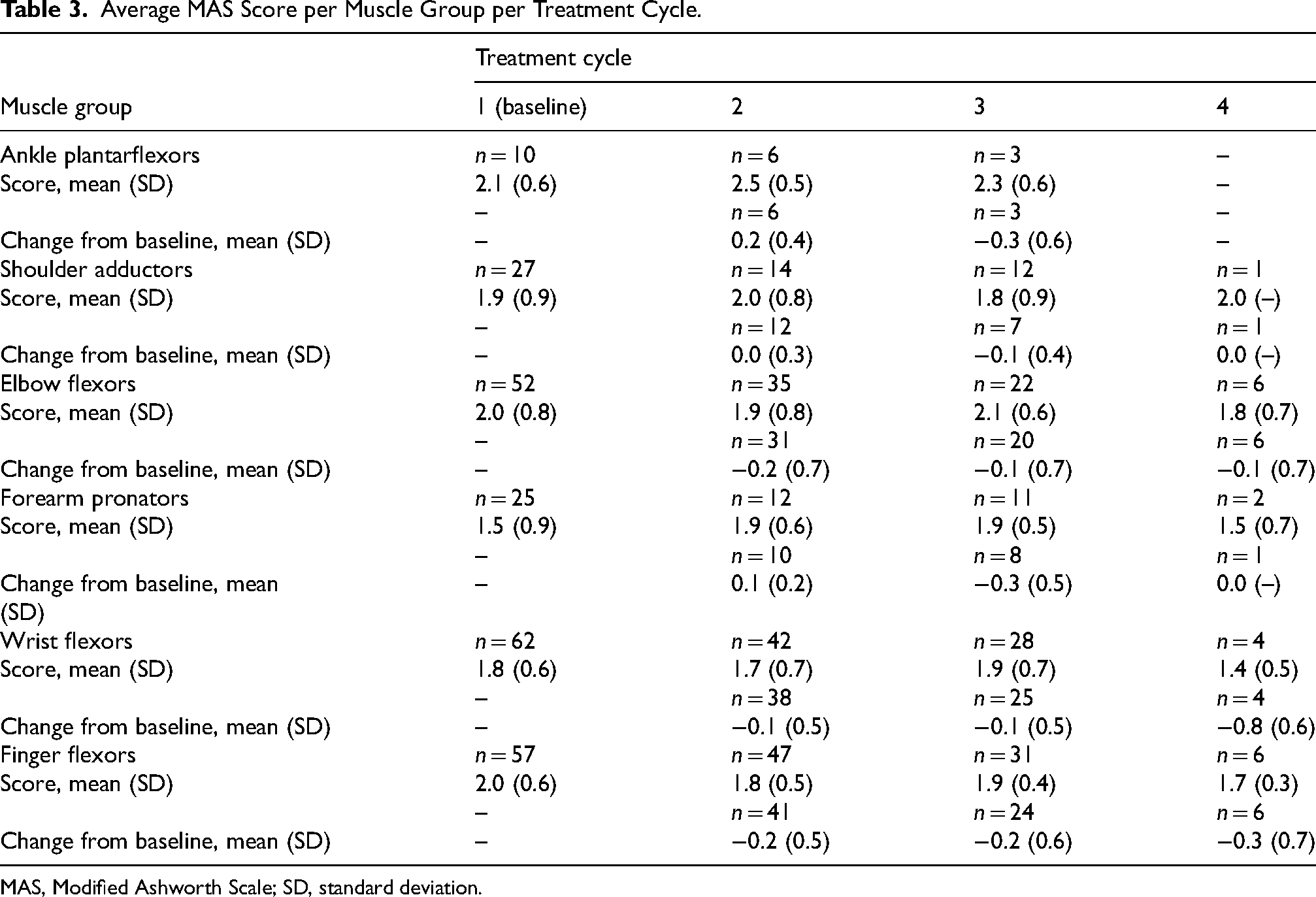
MAS, Modified Ashworth Scale; SD, standard deviation.
Safety
Of the 121 patients included in the safety population, 7 (5.8%) experienced a total of 10 AEs, 7 of which were classified as serious. One patient (0.8%) discontinued treatment due to an AE, and the same patient had an AE that resulted in death. None of the reported AEs were considered to be related to aboBoNT-A treatment. The most common class of AE was nervous system disorders, affecting three patients (2.5%; cerebrovascular accident, subarachnoid hemorrhage, and syncope) (Table 4). Special situations recorded throughout the observation period included off-label use in 10 patients (8.3%) and overdose in 13 patients (10.7%), neither of which were associated with any AEs.
Incidence of Adverse Events in the Safety Population (N = 121).
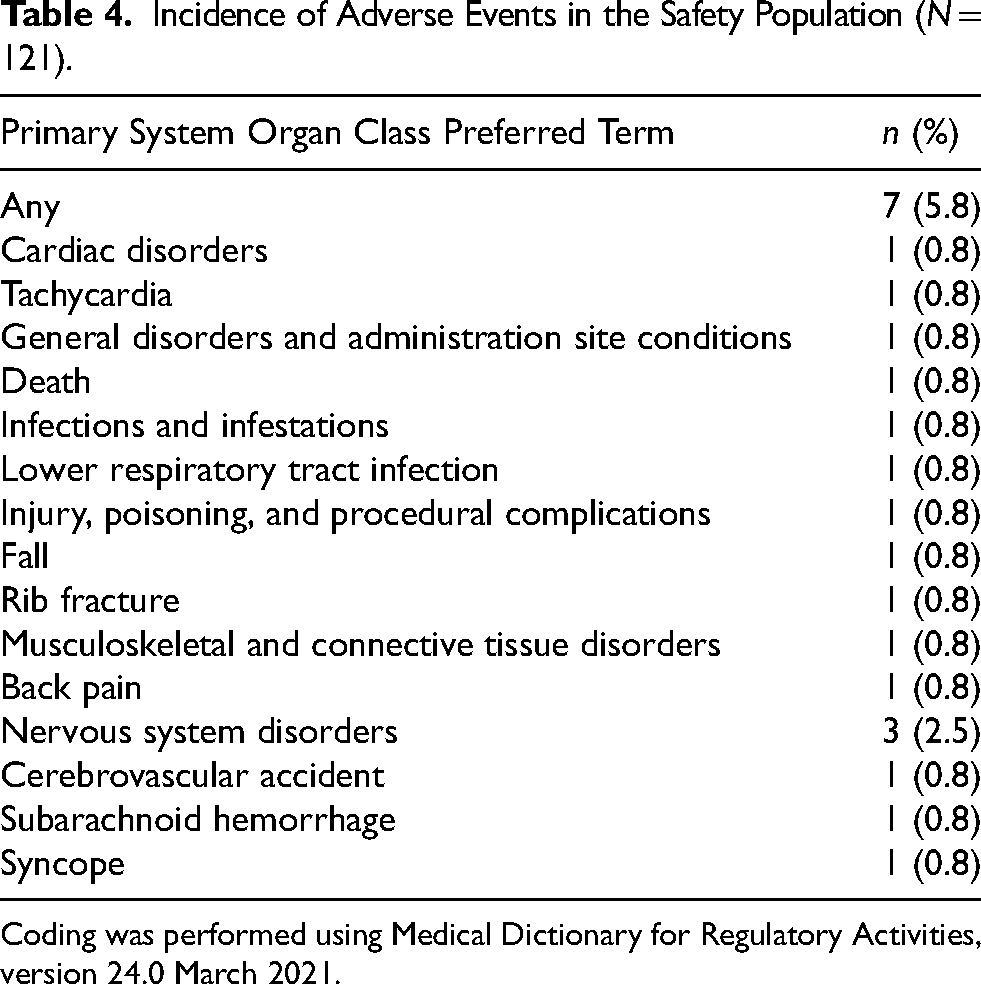
Coding was performed using Medical Dictionary for Regulatory Activities, version 24.0 March 2021.
Discussion
This multicenter, retrospective study aimed to address the lack of information regarding the routine clinical use of aboBoNT-A for the treatment of patients with adult focal spasticity in the UK. In total, 108 patients were included in the study population, and baseline characteristics indicated that most patients had chronic spasticity. The primary objective of the study was to describe the dose and injection interval of aboBoNT-A with single and repeated injection cycles for the treatment of adults with focal ULS and/or LLS. The median interval between diagnosis of neurological condition and first aboBoNT-A injection exceeded 3.5 years; this may have been impacted by the inclusion of patients with lifelong conditions such as cerebral palsy, which is typically diagnosed between 1 and 2 years of age (Novak et al., 2017).
The mean total dose administered across all injection cycles was lower than the recommended maximum dose of 1000 U for ULS and 1500 U for LLS, or ULS and LLS combined (Electronic Medicines Compendium, 2023a, 2023b). However, this does not necessarily suggest suboptimal use because the recommended dose varies between specific muscles, and guidelines state that the lowest effective dose should be administered (Electronic Medicines Compendium, 2023a, 2023b). Only patients treated in line with the aboBoNT-A SmPC were included in this study, with approximately 50% of patients excluded at the pre-screening stage, often due to the use of a dose that was higher than recommended (either per muscle or cumulative) or incomplete data verifying inclusion eligibility. Therefore, the mean total dose used in routine clinical practice may be higher than the value reported in this study. Previous studies that included patients who received off-label treatment reported higher total doses than we have reported here. In a survey of 275 European healthcare professionals, the mean total dose ranged from 508 U to 773 U for ULS and 600 U to 832 U for LLS (Hubble et al., 2013). These values are consistent with the findings of a single-center, retrospective study that included 54 patients who received a mean total dose of 753.7 U at first injection and 703.3 U at re-injection (ClinicalTrials.gov, 2021).
The mean time interval between aboBoNT-A injections was approximately 5 months (∼22 weeks), slightly exceeding the expected injection interval of 16–20 weeks. The main reasons for re-injection were recurrence of symptoms or to maintain symptom control, although there was variability between patients treated for ULS and LLS and between injection cycles. The extended interval between injections may reflect variability in the scheduling of planned follow-up visits in routine clinical practice, which in turn may affect outcome measures, as the results from assessments that have been delayed may show less of a treatment effect. There may also have been selection bias as only patients who had ≥12 weeks between injections (in line with the SmPC) were included. A real-world study evaluating routine clinical practice in Poland included data from 108 adults with ULS and observed similar mean (SD) injection intervals of 4.4 (1.4) and 4.5 (1.2) months between the first and second visits and the second and third visits, respectively (Sarzyńska-Długosz et al., 2020). The authors suggested that this may have been to reduce burden for both patients and physicians, as the timing of re-injection is influenced by multiple factors including capacity constraints, patient illness, availability of transport, and diversity in routine treatment practice (Sarzyńska-Długosz et al., 2020). Findings from our study are consistent with larger international studies, such as the ULIS-III prospective, observational cohort study, in which the mean (SD) injection interval of aboBoNT-A injections for ULS across the study was approximately 6 months (∼188 days) (Turner-Stokes et al., 2021). Similarly, the AboLish prospective, longitudinal, observational cohort study for aboBoNT-A injections for LLS reported the mean (SD) injection interval across the study was 18.3 (6.1) weeks (Esquenazi et al., 2024).
Data from this study further support aboBoNT-A being a well-tolerated and effective treatment for adult focal spasticity. The safety profile was consistent with the SmPC and other previously published data (ClinicalTrials.gov, 2021; Electronic Medicines Compendium, 2023a, 2023b). Methods for assessing effectiveness varied, and GAS, MAS, and Tardieu Scale were recorded for a minority of patients, which limited the ability to fully evaluate aboBoNT-A effectiveness. MAS was used more frequently than the Tardieu Scale, despite the latter being reported as a more appropriate clinical measure of spasticity (Haugh et al., 2006), which is likely to reflect the time constraints as Tardieu Scale assessments take longer to perform. Facilitating the measurement of treatment outcomes by allocating adequate time and resource should be encouraged as the results can inform treatment decisions leading to greater efficiency in clinics and an improved patient journey.
Limitations
A limitation of this study is the failure of ∼50% of screened patients to be eligible for inclusion. Based on investigator feedback, most of these patients were excluded at the pre-screening stage due to off-label use; therefore, the real-world patient population treated with aboBoNT-A in routine clinical practice in the UK remains elusive, and this has implications from a commissioning perspective. The results of this study may not be representative of the wider patient population with focal ULS and/or focal LLS treatment with aboBoNT-A in routine clinical practice in the UK. In addition to the use of a dose that was higher than recommended, as previously discussed, patients were commonly excluded from the study due to injection into off-label muscles or treatment for off-label indications. In particular, the limited number of lower limb muscles recommended for injection in the SmPC meant that the number of patients with LLS included in the study population was relatively low as patients with injections in the hamstrings, adductors, and hip flexors were excluded (Electronic Medicines Compendium, 2023a, 2023b). These findings suggest that further studies into aboBoNT-A dosing are warranted to provide evidence to support an update to the SmPC that would permit its use in additional muscles. Other limitations of this study are inherent to its retrospective nature, specifically that the quality of data was reliant on the accuracy and completeness of patient hospital records, and the number of patients with missing data varied for different endpoints. Documentation practices across the sites also meant that a proportion of patients had insufficient data to evaluate inclusion eligibility. Although the eCRF (Supplementary Table S1) included data and outcomes that were most likely collected by centers, there was variability in the quality of individual patient data entered between centers, and patient goals were not methodologically recorded across all centers.
Conclusions
This study provides a much-needed insight into the use of aboBoNT-A for adult focal spasticity in the UK, and the findings can be used to support clinical practice and inform commissioning decisions related to the use of aboBoNT-A in this indication. Additionally, as this analysis is based on data that were collected shortly before the onset of the COVID-19 pandemic, it may serve as a useful comparator in future studies investigating the impact of the pandemic on treatment practices.
Supplemental Material
sj-docx-1-nre-10.1177_10538135251325755 - Supplemental material for A Multicenter, Retrospective, Real-World Study of Treatment Outcomes Following on-label AbobotulinumtoxinA Injections for Adult Focal Spasticity in the United Kingdom
Supplemental material, sj-docx-1-nre-10.1177_10538135251325755 for A Multicenter, Retrospective, Real-World Study of Treatment Outcomes Following on-label AbobotulinumtoxinA Injections for Adult Focal Spasticity in the United Kingdom by Moheb Gaid, Caroline Brown, Mario Ippolito, Vadim Degtiar, Pascal Maisonobe and Samantha Orridge in NeuroRehabilitation
Footnotes
Acknowledgments
The authors thank all patients involved in the study, and their caregivers and care team, and investigators and research staff in participating institutions. The authors thank Rosie Pryor, PhD, and Pallavi Patel, PhD, CMPPTM of Luna, OPEN Health Communications (London, UK) for providing medical writing support that was sponsored by Ipsen (Slough, UK) in accordance with Good Publication Practice guidelines.
Ethics Approval and Consent to Participate
This study adhered to all local regulatory requirements applicable to non-interventional studies and received approval from the UK National Health Service Health Research Authority and Health and Care Research Wales (research ethics committee reference 20/HRA/5179, October 19, 2020). Patient data were pseudo-anonymized prior to transfer to external researchers for analysis with confidentiality preserved at all times, therefore patient consent was not required.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was sponsored by Ipsen. Ipsen was responsible for the design of the study protocol and the statistical analysis plan.
Declaration of Conflicting Interests
MG: Travel support & funding to institution–Ipsen; CB: Consulting & honoraria payment, & travel support–Ipsen; MI, PM, VD: Employees of Ipsen; SO: Research funding–Ipsen, training funding–Merz Pharma.
Data Availability
Qualified researchers may request access to patient-level study data that underlie the results reported in this publication. Additional relevant study documents, including the clinical study report, study protocol with any amendments, annotated case report form, statistical analysis plan, and dataset specifications may also be made available. Patient-level data will be anonymized, and study documents will be redacted to protect the privacy of study participants.
Where applicable, data from eligible studies are available 6 months after the studied medicine and indication have been approved in the USA and European Union or after the primary manuscript describing the results has been accepted for publication, whichever is later.
Further details on Ipsen's sharing criteria, eligible studies, and process for sharing are available here (https://vivli.org/members/ourmembers/). Any requests should be submitted to ![]() for assessment by an independent scientific review board.
for assessment by an independent scientific review board.
Supplemental Material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.