
Abstract
The Serratia marcescens complex contains important opportunistic pathogens of humans and vertebrate animals, as well as insects and other invertebrates. To date, the methods used for the identification of species within the genus Serratia, including PCR assays, have poor discriminatory power and may require further molecular typing or genomic sequence analysis to determine clinical relevance. We developed a duplex TaqMan probe–based quantitative real-time PCR (qPCR) assay targeting the chiP gene, which is involved in chitin degradation and transport, and the ureD gene, which is involved in urease production. This allowed us to simultaneously identify all members of the S. marcescens complex (chiP positive) and differentiate those most likely to act as insect pathogens (chiP and ureD positive). We applied our assay to identify potentially entomopathogenic members of the S. marcescens complex in the context of a conservation program for the critically endangered insect Dryococelus australis and found it to be a useful aid for rapid and accurate detection of infection with S. marcescens complex strains in insects and determination of their clinical relevance. By targeting 2 genes likely to be virulence factors, this assay may also be of use for research investigating the pathogenesis of entomopathogenic Serratia spp.
In 2001, a small remnant population of Lord Howe Island stick insects (Dryococelus australis Montrouzier) was rediscovered, ~80 y after the species was believed to have been extirpated following the incursion of rodents onto Lord Howe Island, Australia. 47 Several insects were collected from a nearby islet, Ball’s Pyramid, to establish a captive breeding program at Melbourne Zoo in Australia. 27 This program is now 20-y-old and contains ~500 individuals.7,28 One of the most significant challenges to the continued success of the captive breeding program for D. australis has been the impact of infections caused by bacterial entomopathogens.5,11 These pathogens have resulted in disease outbreaks, elevated baseline mortalities, and difficulties in establishing additional captive insurance populations in other institutions outside of Melbourne Zoo.
The most common microorganisms isolated from dead and moribund captive D. australis are members of the Serratia marcescens complex,1,18 which includes important opportunistic pathogens of humans and domestic animals, 24 most commonly as nosocomial infections.2,15,32 The S. marcescens complex forms a group of genetically related taxa that contains the species S. marcescens sensu stricto, S. ureilytica, S. nematodiphila, S. surfactantfaciens, S. nevei, and S. bockelmannii.12,43 Microbiologic and histopathologic investigations of infected D. australis have indicated the important role of this complex as likely pathogenic agents, and although the Serratia spp. present in the environment of the captive insects are genetically diverse, the most prominent and persistent strains associated with D. australis mortalities belong to an insect-associated clade of the S. marcescens complex.1,5 An insect infection study using Apis mellifera demonstrated the pathogenic potential of a representative strain, S. ureilytica AM923, in insects. 1
There have been many taxonomic revisions within the genus Serratia, but recent advances in genomic sequence analysis have allowed a more comprehensive description of the genus. 63 The S. marcescens complex is phylogenetically distinct from other species in the genus.1,43 A second group of closely related organisms in the genus, the S. liquefaciens complex, contains S. liquefaciens sensu stricto, S. proteamaculans, S. quinivorans, and S. grimesii. 24 The species within both of these groups are closely related and difficult to distinguish using conventional microbiologic methods or 16S rRNA sequencing, but they belong to distinct clades that are associated with specific ecological niches and have differing pathogenic potential for different hosts.43,63 Some clades are almost exclusively isolated from clinical human infections and nosocomial sources, whereas others are more commonly associated with environmental sources or insects.1,43
Identification of Serratia spp. from infections has traditionally relied upon extensive testing of growth and biochemical characteristics, which is time-consuming and labor-intensive.21,24 Molecular methods have been used to detect clinical S. marcescens infections more rapidly and to trace outbreaks caused by S. marcescens in hospitals and other care settings.48,54 In clinical and research applications, the molecular identification of Serratia spp. has primarily relied upon PCR assays targeting highly conserved regions, such as the gyrB gene 49 or 16S rRNA.30,64 However, these assays must be used in combination with other specialized methods, such as DNA fingerprinting techniques or genomic sequencing and analysis, to distinguish between closely related strains and species.38,44,65
Although studies investigating Serratia spp. infections in invertebrates have used PCR assays developed for clinical human isolates of S. marcescens,19,31 recent genomic studies indicate that most insect-associated strains are in a clade distinct from most human-associated strains.1,43 Therefore, assays developed for clinical applications may be suboptimal for investigating Serratia spp. infections in insects, and there is a need for more specific assays. Previous work demonstrated that some gene targets, such as those encoding for chitinases and nucleases, are widely conserved across the S. marcescens complex. 1 Another interesting target is the urease gene operon, which is found in all strains from the insect-associated clade, but is rarely found in other clades of the S. marcescens complex. 1 Our objective was to develop multiplex TaqMan probe–based quantitative real-time PCR (qPCR) assays to develop a sensitive and specific rapid test that could identify isolates of the S. marcescens complex from infections in insects and accurately distinguish insect-associated members of the S. marcescens complex from isolates more likely to be associated with humans or the environment.
Materials and methods
Bacterial isolation and identification
Swab samples from the hemocoel and gut contents of recently deceased D. australis from Melbourne Zoo were immersed in Amies transport medium (Copan) and stored at 4°C until processing. These samples were inoculated onto sheep blood agar (SBA; 5% blood cells) and MacConkey agar (Thermo Fisher), which were incubated overnight at 37°C. We then subcultured each isolate on SBA from individual colonies picked from the primary plates and identified the isolates using conventional microbiologic identification methods (morphology and biochemical tests), as described, 29 and tested them for the production of urease. Pure cultures from each isolate were stored in tryptone soy broth and 30% glycerol at −80°C.
Culture databases
We established a database of 100 bacterial isolates from insect, animal, and environmental sources at the Clinical Microbiology Laboratory of the Melbourne Veterinary School, University of Melbourne (Suppl. Table 1). The collection included Serratia spp. and non-Serratia spp. identified using conventional identification methods, commercial kits (API; bioMérieux), 16S rRNA gene sequencing, biotyping to distinguish closely related Serratia spp., 18 or whole-genome sequencing and analysis.1,2 We tested all isolates for the production of urease by inoculation onto urea agar slopes and incubation at 37°C for 72 h.
We also established a database of microorganisms cultivated from the gut and celomic swabs of phasmids at Melbourne Zoo between March 2019 and April 2022 (Suppl. Table 2). The isolates from stick insects were identified to the genus level using conventional identification methods, the EnteroPluri-Test system (Liofilchem), API kits (bioMérieux), or 16S rRNA gene sequencing for organisms that could not be identified using standard conventional methods.
DNA extraction
We extracted whole DNA from pure cultures (1.5 mL) grown in tryptone soy broth (Thermo Fisher) after 18 h incubation at 37°C using the Wizard genomic DNA (gDNA) purification kit (Promega) and standardized the sample DNA concentrations to 50 ng/µL for use in the assays. To obtain crude DNA templates from culture, we suspended 1–3 bacterial colonies picked from agar plates in sterile saline (100 µL) and lysed them by incubation at 100°C for 3 min.
Target gene assessment
We generated a genomic sequence dataset from all complete, scaffold, or contig assemblies from the NCBI RefSeq database (downloaded 2021.04.23, https://www.ncbi.nlm.nih.gov/refseq/) in the genus Serratia, and collated the metadata for all sequences from the file headers. We identified the clonal and duplicated genomes by examining their metadata, verified them by referencing their full records in NCBI, and removed them from the dataset, as well as the genomes of S. symbiotica and S. microhaemolytica. We then systematically re-annotated all genomes with the prokaryotic software annotation tool Prokka v.1.14.6. 51 We extracted the full set of coding sequences (CDSs) from these genomes and searched them for specific CDSs, using a selection of annotated sequences from S. ureilytica AM923 as queries. Searches were performed using Nucleotide BLAST (BLASTn; https://blast.ncbi.nlm.nih.gov/Blast.cgi?PAGE_TYPE=BlastSearch) to identify matches with ≥70% identity, 63 and 90% query coverage. For S. marcescens complex–specific PCR targets, we used the AM923 chiA (chitinase A), chiP (chitoporin), chb (chitobiase), and nucA (nuclease) genes as queries. To assess PCR targets for insect-associated strains of the S. marcescens complex, we used ureA2 (urease subunit gamma 2), ureB (urease subunit beta), ureC (urease subunit alpha), ureE (urease accessory protein UreE), ureF (urease accessory protein UreF), ureG (urease accessory protein UreG), ureD (urease accessory protein UreD), and hoxN (high-affinity nickel transport protein). We used CD-HIT v.4.8.122,37 to consolidate the identical gene sequences into unique clusters, compiled these unique sequences into multi-FASTA files, and created multiple alignments for each of the multi-FASTA files using MUSCLE v.3.8.1551. 20 We used FastTree v.2.1.11 with the -gtr parameter selected45,46 to infer single gene phylogenetic trees and visualized each of the trees using MEGA v.11. 61 Finally, we performed BLAST searches (https://blast.ncbi.nlm.nih.gov/Blast.cgi) with each of the gene sequences using the CDSs from AM923 as queries against the NCBI database with Serratia excluded to check for matches to non-Serratia spp.
Primer design
Once candidate genes were chosen as potential targets for the qPCR assays, we created consensus sequences from the multiple alignments in Geneious Prime v.2021.1.1 using the Generate Consensus Sequence tool with a 95% threshold (Biomatters). We then designed primer and internal oligonucleotide sets in Primer3Plus v.3.1.0 (https://www.primer3plus.com/index.html). We evaluated the in silico values and penalty scores from Primer3Plus for each set of primers and internal oligonucleotides that were generated, and selected primer sets with the lowest probabilities for self-complementarity, mispriming, and secondary structure formation. We then conducted BLAST searches with the selected primer sets against both the curated Serratia database and BLAST with Serratia excluded. Lastly, the sequences of reference genomes for species within and outside the genus Serratia were downloaded from the NCBI RefSeq database and the chosen primer sets compared to the corresponding genes in Geneious Prime using the Multiple Align tool with a Geneious alignment and default settings.
Assay optimization
We used gDNA from S. ureilytica AM923 to establish positive control standards of 5–5 × 105 DNA copies/µL. PCRs were conducted using 250 nM TaqMan probes, 1× Brilliant II QPCR master mix (Agilent), 30 nM reference dye, and gDNA template (2 µL). The qPCR assays were incubated in a real-time PCR cycler (Rotor-Gene Q; Qiagen) and analyzed using the Rotor-Gene Q v.2.3.1.49 (Qiagen). Reaction conditions were an initial denaturation incubation at 95°C for 10 min, then incubation through 40 cycles of denaturation at 95°C for 30 s, and annealing/extension at 60°C for 60 s, with fluorescence measured during the annealing/extension step of each cycle. All standard assays were conducted in triplicate. Once optimal parameters were determined, we compared the standard curve produced by the multiplex qPCR to the singleplex qPCRs to ensure that the reproducibility, linearity, sensitivity, and reaction efficiency of the assay were not significantly affected by multiplexing.
Establishing limits of detection
To establish the limits of detection (LODs) for the qPCR assays, we used an overnight incubated culture of S. ureilytica AM923 in tryptone soy broth at an OD600 of 0.5 to make serial 10-fold dilutions and performed viable cell counts to determine the concentration of bacteria. We then lysed samples of the broth dilutions by incubating at 100°C for 3 min to prepare crude DNA templates for the qPCR assays.
Validation and application of qPCR assays
To validate our qPCR assays, we first extracted DNA from the cultures of known bacterial isolates (Suppl. Table 1), standardized all gDNA samples to 50 ng/µL, and conducted multiplex qPCR assays. In addition, we used crude DNA templates from the cultures of isolates from captive phasmids (Suppl. Table 2) for multiplex qPCR assays. Lastly, we applied the multiplex qPCR assays to crude DNA templates from cultures obtained from swabs of recently deceased D. australis from the quarantine enclosures at Melbourne Zoo.
Results
Distributions of target genes
To investigate potential gene targets for qPCR assays that would identify isolates in the S. marcescens complex and distinguish insect-associated strains, we compared the annotated sequence data for S. ureilytica AM923 1 to a dataset of 950 unique assemblies from the genus Serratia. BLASTn searches against the dataset using the CDSs from AM923 as queries found that each of the genes chiA, chiP, chb, and nucA were present in >90% of the Serratia spp. sequences, but that chb and chiP were present in >95% of sequences, including members of the S. marcescens complex, S. liquefaciens complex, S. plymuthica, S. odorifera, S. fonticola, and S. ficaria. BLASTn searches with each of the genes in the urease operon only identified members of the S. marcescens complex.
Primer evaluation
We targeted a 78-bp region at the start of the chiP gene as a S. marcescens complex–specific PCR, and a 120-bp region of the ureD gene as an insect-associated S. marcescens complex–specific PCR (Table 1). The multiple alignment of the chiP primer set against reference sequences within and outside the genus Serratia found they had 100% identity with S. ureilytica, S. marcescens, and S. liquefaciens, a single mismatch in the probe with the more distantly related species S. ficaria, and poor predicted binding to non-Serratia genomes containing the chiP gene (Fig. 1A). The ureD primer set had 100% identity with the reference sequences from the S. marcescens complex (S. ureilytica, S. nevei, and S. surfactantfaciens) but poor predicted binding to sequences from the closely related genus Yersinia (Fig. 1B).
Primers and probes selected for qPCR assays to identify the chiP and ureD genes of Serratia.

Both oligonucleotide probes contained a fluorescent reporter (HEX or 6-FAM), internal quencher (ZEN), and 3′ quencher (Iowa Black FQ).
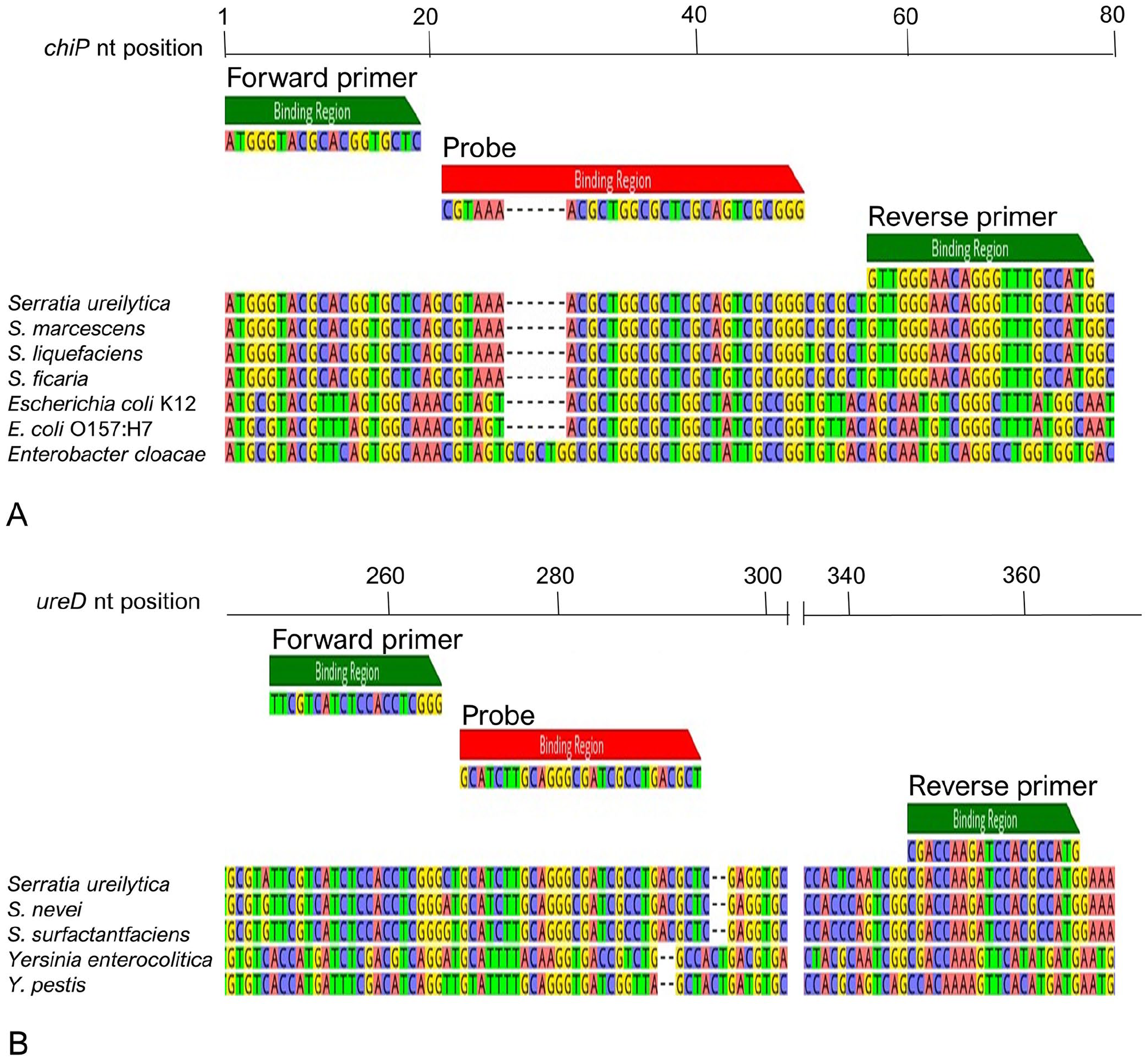
Primer sets aligned to the corresponding gene sequences from reference genomes for Serratia spp. and non-Serratia spp. containing the (
Optimization of qPCR assays
We determined that primer concentrations of 300 nM for both the chiP and ureD singleplex qPCR assays yielded sensitive detection of S. ureilytica AM923. The assays demonstrated optimal analytical sensitivity and specificity using a fluorescence threshold of 0.15 for the chiP singleplex qPCR and 0.12 for the ureD singleplex qPCR (Suppl. Fig. 1). The assays produced standard curves with a linear dynamic range of 103–107 copies/reaction, high repeatability as indicated by the SDs of the mean Ct values (Suppl. Table 3), and amplification efficiencies of 94% for the chiP assay and 95% for the ureD assay.
Multiplexing the reaction did not affect the performance of either of the assays substantially, with linearity and dynamic ranges for both targets similar to those for the singleplex assays (Fig. 2), and amplification efficiencies of 95% for the chiP target and 91% for the ureD target. Mean Ct values and their SDs were highly similar to those of the singleplex assays when applied to standardized concentrations of AM923 gDNA, indicating similar analytical sensitivity and repeatability between the singleplex and duplex assays (Suppl. Tables 3, 4).
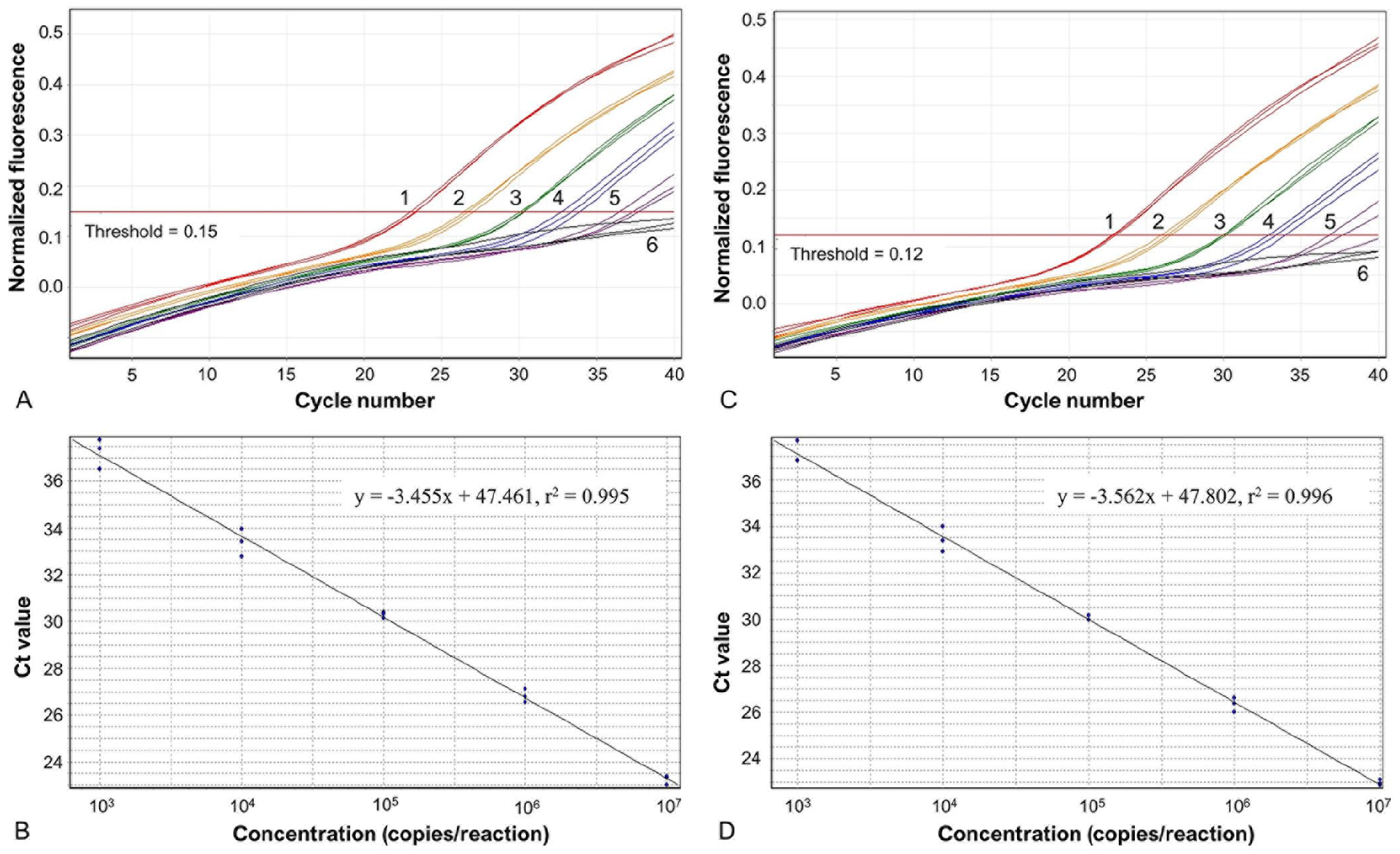
Duplex qPCR assay for the Serratia chiP and ureD targets with standard curves for S. ureilytica AM923 gDNA. Amplification plots for the Serratia (
Analytical sensitivity and specificity
The LODs for the multiplex qPCR assay were ~10 cfu per reaction for both the chiP and ureD targets when using pure cultures (Suppl. Fig. 2), with highly similar Ct values obtained in each of the 2 assays (Suppl. Table 5).
We tested our duplex qPCR assay on a collection of bacterial isolates and obtained positive results in the chiP assay for all 56 isolates from the S. marcescens complex, as well as the single isolate from the S. liquefaciens complex (Table 2). The single isolate identified as S. rubidaea and all non-Serratia isolates (n = 42) were negative in the chiP assay. Only urease-producing isolates of the S. marcescens complex (n = 34) were positive in the ureD assay, and all non-Serratia bacterial species, including those that were urease producers, were negative (Table 2). In addition, when the duplex qPCR assay was applied to a collection of isolates cultured from the celomic and gut swabs of anesthetized and/or recently deceased stick insects, all of the urease-positive cultures of Serratia spp. (n = 19) were positive in both the chiP and ureD assays, all urease-negative Serratia spp. (n = 12) were positive in the chiP assay only, and all other non-Serratia isolates (n = 60) were negative in both assays (Table 3).
Performance of duplex chiP and ureD qPCR assay when used to test gDNA extracted from cultures of characterized bacterial isolates.
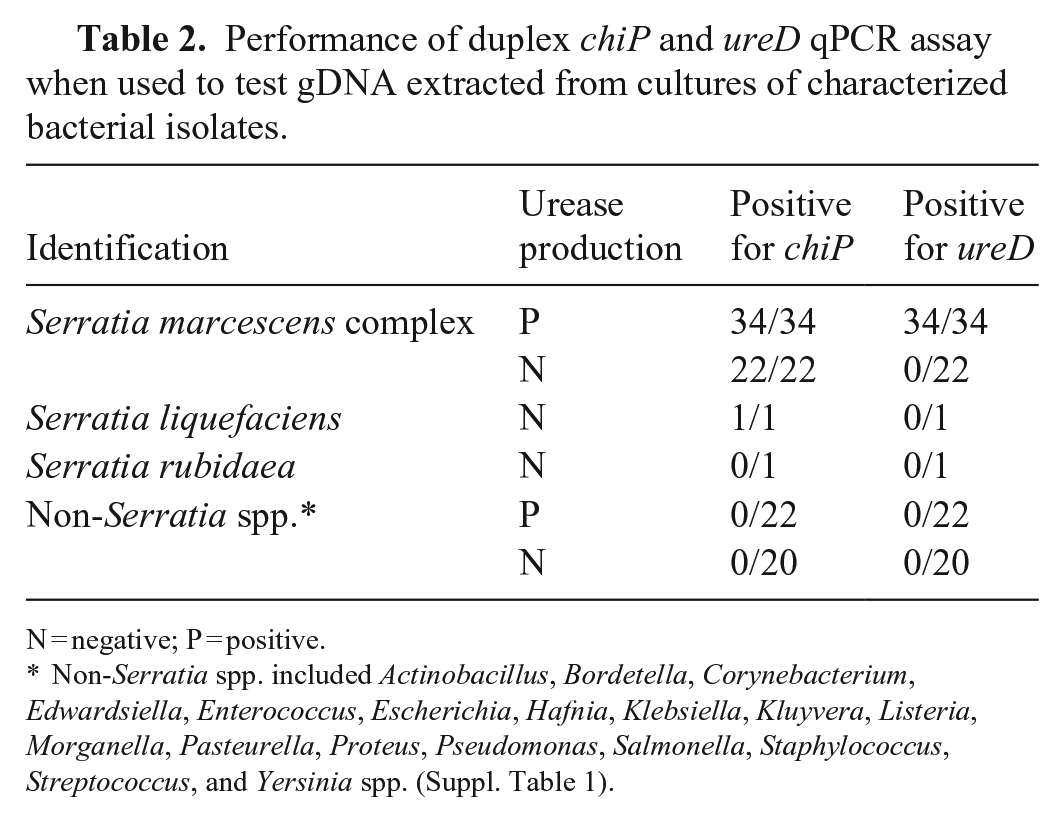
N = negative; P = positive.
Non-Serratia spp. included Actinobacillus, Bordetella, Corynebacterium, Edwardsiella, Enterococcus, Escherichia, Hafnia, Klebsiella, Kluyvera, Listeria, Morganella, Pasteurella, Proteus, Pseudomonas, Salmonella, Staphylococcus, Streptococcus, and Yersinia spp. (Suppl. Table 1).
Performance of duplex chiP and ureD qPCR assay when used to test cultures from swabs of dead or anesthetized Dryococelus australis and Acrophylla wuelfingi at Melbourne Zoo, 2019–2022.
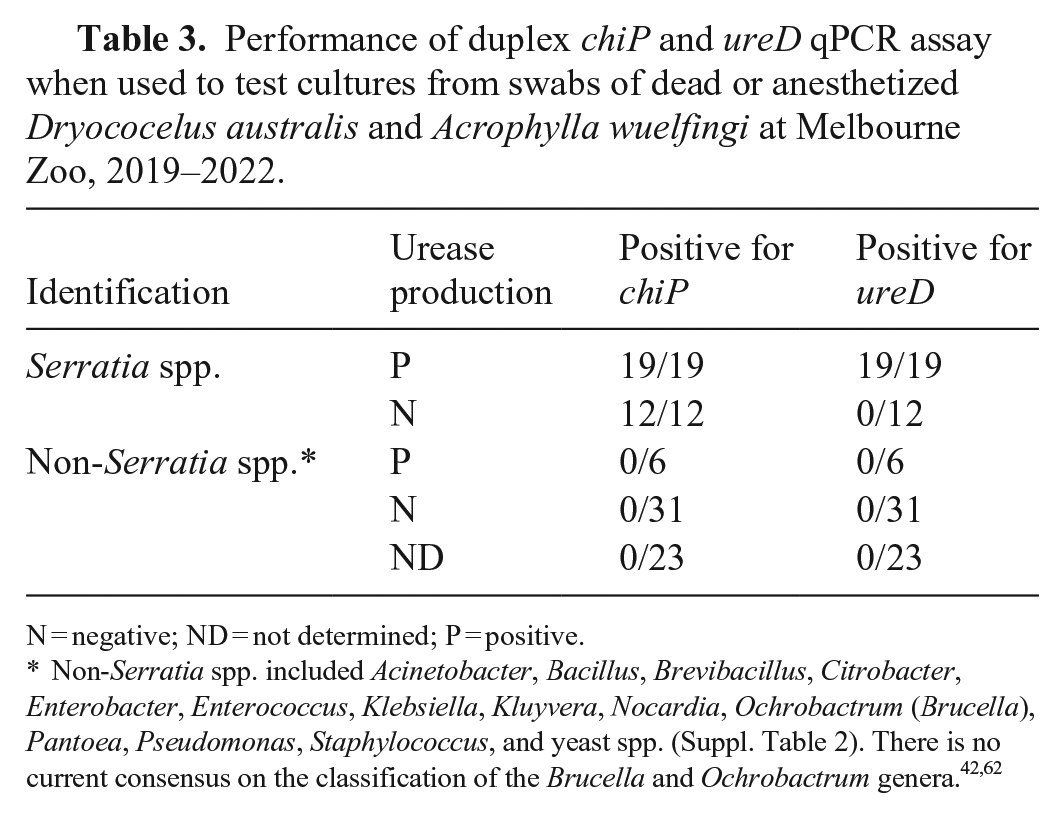
N = negative; ND = not determined; P = positive.
Non-Serratia spp. included Acinetobacter, Bacillus, Brevibacillus, Citrobacter, Enterobacter, Enterococcus, Klebsiella, Kluyvera, Nocardia, Ochrobactrum (Brucella), Pantoea, Pseudomonas, Staphylococcus, and yeast spp. (Suppl. Table 2). There is no current consensus on the classification of the Brucella and Ochrobactrum genera.42,62
Application of the assay in a captive insect population
We used the duplex qPCR assay to test D. australis in the quarantine enclosures at Melbourne Zoo, where Serratia spp. had not been identified previously. When suspected isolates of Serratia spp. were cultured from the celom and gut swabs of recently deceased insects in the quarantine enclosures, we used the duplex qPCR assay to rapidly determine whether the isolates were from the S. marcescens complex and, if they were, whether they were more likely to be insect-associated or environmental strains of the S. marcescens complex. Some cultures were positive for the chiP qPCR target, but none were positive for the ureD target (Suppl. Fig. 3). Subsequent conventional microbiologic identification confirmed that the isolates positive in the chiP qPCR assay were urease-negative members of the S. marcescens complex and that the isolates that were not positive in the chiP qPCR assay were non-Serratia isolates (Table 4).
Performance of duplex chiP and ureD qPCR assay when used to test cultures of swabs of dead or anesthetized Dryococelus australis from the quarantine enclosures at Melbourne Zoo, February and March, 2022.

Identifications based on conventional identification methods and EnteroPluri (Liofilchem) testing.
Discussion
We developed a duplex TaqMan probe–based qPCR assay for the identification and differentiation of members of the S. marcescens complex infecting or colonizing insects. We selected the Serratia chiP gene, encoding the S. marcescens chitoporin (SmChiP), as the target for a qPCR assay that could identify all members of the S. marcescens complex. Chitoporins have been described in Vibrio spp., 33 Escherichia coli, 57 and S. marcescens, 3 and allow the importation across the bacterial outer membrane of the degradation products of chitin generated by chitinolytic enzymes, chitooligosaccharides. These degradation products are then used as nitrogen and carbon sources. 56 Chitin is an essential component of both the cuticle of insect exoskeletons and the peritrophic membrane, a protective layer lining the digestive tracts of many insects. 35 Chitinases from entomopathogenic bacteria, such as S. marcescens, can cause degradation and perforation of the peritrophic membrane, leading to decreased feeding, weight loss, and mortality from septicemia. 34 Investigations of mortalities in captive D. australis found histopathologic evidence of gram-negative bacteria penetrating through the gut lining of affected insects and entering the hemocoel. 5 In addition, Serratia spp. were frequently isolated from the normally sterile hemocoel of euthanized or anesthetized D. australis,1,18 so it is likely that the degradation of chitin in the peritrophic membrane plays an important role in the pathogenesis of disease caused by these organisms.
The chiP gene should be a useful target not only for identifying most entomopathogenic strains of the S. marcescens complex, but also most clinically relevant strains of Serratia spp. in humans and animals. BLAST searches found that the genomes of all species in the S. marcescens and S. liquefaciens complexes contained chiP, as well as the more distantly related species S. fonticola, S. odorifera, S. plymuthica, and S. ficaria. The vast majority of Serratia-associated clinical disease in vertebrate and invertebrate animals, as well as nosocomial infections in humans, are caused by members of the S. marcescens complex, members of the S. liquefaciens complex, or S. fonticola.24,39 We tested our qPCR assay against a range of isolates from insects, the environment, and clinical cases in domestic animals, and yielded positive results for the chiP target for all isolates from the S. marcescens complex. To determine the analytical specificity of the test, further validation with a panel of environmental and clinical human isolates would be needed, to ensure the utility of the assay for the entire S. marcescens complex and for other Serratia spp. The single S. liquefaciens isolate in our collection was also positive, as expected based on database searches with the primer sequences, but further validation with more strains from the S. liquefaciens complex, as well as other species across the genus Serratia, would be useful to fully understand the potential applications of the assay. The chiP qPCR is unlikely to identify Serratia species that do not metabolize chitin, such as S. rubidaea, but these species are rarely implicated in clinical cases in humans or animals. 23 Therefore, the chiP target could be used alone or in addition to assays targeting the entire Serratia genus, such as those based on the 16S rRNA gene, 30 gyrB, 49 or luxS, 66 to rapidly determine if Serratia spp. identified using these assays or other methods are likely to be clinically relevant.
In both free-ranging and artificial environments, insects are likely to be regularly exposed to environmental isolates of Serratia spp. in their food, water, and substrate, and, in captive environments, they may also be exposed to human-associated strains of Serratia spp. 25 Therefore, it is necessary to be able to distinguish clades of Serratia spp. that are more likely to be potentially pathogenic in insects from those that they may transiently carry and are not associated with disease. Our previous investigations revealed that the predominant strains affecting captive D. australis were urease-producing members of the S. marcescens complex, and genomic sequence analysis revealed that the urease gene operon was present in the genomes of all strains from the insect-associated clade of the S. marcescens complex, but rare in those of strains in the human and environmental clades. 1
We selected the ureD gene, part of the urease gene operon, as the target for a qPCR assay to identify insect-associated strains of the S. marcescens complex. Urease production was previously thought to be absent or rare for all species in the genus Serratia, 24 but, in 2005, S. ureilytica, a urease-producing species within the S. marcescens complex, was described. 6 Since then, 2 more urease-producing species in the S. marcescens complex, S. surfactantfaciens and S. nevei,12,60 have also been reported. Also, a 2024 evaluation of strains from insects and their environments suggests that additional urease-positive clades may be present in the S. marcescens complex. 18 Ureases are produced by plants, fungi, and bacteria that hydrolyze urea into ammonia and carbon dioxide. 9 They allow plants to convert urea, widely used as a fertilizer in agriculture, into a nitrogen source for plant growth, 55 and urease-producing bacteria, such as Klebsiella aerogenes and S. ureilytica, are able to use urea as an exclusive source of nitrogen.6,40 The production of urease also allows some bacteria to neutralize acids in the highly acidic gastrointestinal tracts of mammals,14,16 and may allow urease-producing members of the S. marcescens complex to survive passage through the acidic environment of the phasmid foregut and anterior midgut. 41
Ureases produced by plants have potent insecticidal activity against a range of insect orders.8,59 They can cross the intestinal epithelium of insects26,36 and cause a range of entomotoxic effects, such as hemocyte aggregation, central neurotoxicity, neuromuscular blockade, diuresis, and reduced gut contractility.4,10,17,58 The entomotoxicity of bacterial ureases has not been extensively investigated in Serratia spp., but most species from the closely related genus Yersinia have strong urease activity, and they have significant entomopathogenic effects on fleas that can be reversed by a urease inhibitor. 13 Yersinia pestis, the causative agent of bubonic plague, has a functional mutation in the ureD gene that prevents it from producing urease and allows it to be transmitted by fleas without causing mortality in its insect host. 50 Therefore, it is tempting to speculate that the presence of a functional urease gene operon in the insect-associated clade of the S. marcescens complex could also play an important role in pathogenesis in insects, but its contribution to disease remains to be clarified.
Our qPCR assay had high analytical sensitivity and specificity when used to identify and differentiate pure cultures, but lower performance is expected for detection from more complex samples, such as swabs, soil, and insect frass. PCR inhibitors in clinical samples can interfere with the sensitivity of qPCR assays, 53 and the unique composition of frass from phytophagous insects such as phasmids and larval lepidopterans may reduce the performance of the reaction.41,52 We plan to investigate the application of this qPCR assay in live insects and concurrently develop alternative methods for detection that are less sensitive to the inhibitors present in insect frass.
When we applied the duplex qPCR assay to a captive population of D. australis that had no previous history of infection with Serratia spp. it allowed us to rapidly and reliably detect S. marcescens complex infections and helped us to determine that the strains identified were more likely to be opportunistic organisms rather than the circulating entomopathogenic strains present in other captive populations at Melbourne Zoo. Our assay can be used to inform decisions about, and develop interventions for, the D. australis conservation program, and could also be used as a tool in other clinical and research settings.
Supplemental Material
sj-pdf-1-vdi-10.1177_10406387241313448 – Supplemental material for Development of a qPCR assay to identify and differentiate insect-associated strains of the Serratia marcescens complex
Supplemental material, sj-pdf-1-vdi-10.1177_10406387241313448 for Development of a qPCR assay to identify and differentiate insect-associated strains of the Serratia marcescens complex by Nicholas P. Doidge, Joanne L. Allen, Rhys Bushell, Michael Lynch, Glenn F. Browning and Marc S. Marenda in Journal of Veterinary Diagnostic Investigation
Footnotes
Acknowledgements
We acknowledge the tireless work over 2 decades by the invertebrate, veterinary, and Wildlife Conservation and Science teams at Melbourne Zoo and Zoos Victoria to make the conservation program for Dryococelus australis a success. We also thank Martins Olaogun, Rebecca Agnew, and Dami Omotainse from the Asia-Pacific Centre for Animal Health at the Werribee campus of the Melbourne Veterinary School, University of Melbourne, for their technical expertise and assistance with troubleshooting during the development of our assays.
Part of the material included in this article was presented at the Wildlife Disease Association International Conference on 2022 Jul 26 (Madison, WI, USA), the Veterinary Pathology Congress on 2022 Oct 12 (Melbourne, Victoria, Australia), and the Australian Association of Veterinary Laboratory Diagnosticians Meeting on 2022 Nov 18 (Launceston, Tasmania, Australia).
Declaration of conflicting interests
The authors declare no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.