
Abstract
African swine fever (ASF) is a high-consequence transboundary animal disease caused by African swine fever virus (ASFV). Given that vaccines are not widely available, ASFV detection, including by molecular and serologic assays, is paramount to efficacious control and mitigation of ASF. ASFV-specific antibodies can be detected as early as 7–10 d postinfection in infected animals and may persist for several months or longer. Accurate detection of ASFV-specific antibody is critical for the identification of chronically infected, subclinically infected, or recovered animals. ELISAs are commonly used for the rapid screening of large numbers of animals for ASFV antibodies. The World Organisation for Animal Health recommends that ELISA-positive results should be confirmed with a second serologic method, such as an indirect immunofluorescent assay, indirect immunoperoxidase test (IPT), or immunoblot test. Commercial kits are not available for those tests. We developed and validated an in-house IPT by using a currently circulating genotype II ASFV strain as antigen. The sensitivity and specificity of the in-house IPT are comparable to the reference IPT developed by an international ASFV reference laboratory and superior to a commercial blocking ELISA.
African swine fever (ASF) is a complex and high-mortality viral disease that affects all breeds of swine (Sus scrofa), including wild boars and domestic pigs. The etiology of ASF is African swine fever virus (ASFV; Asfarviridae, Asfivirus), a double-stranded, double-enveloped DNA virus. ASFV has been classified into different genotypes based on the B646L gene (coding structural protein p72) 9 or different hemadsorption inhibition (HAI) groups. 6 However, it is believed that ASFV has only one serotype. The current ASF outbreak, caused by a genotype II ASFV, 3 started in 2007 8 in the Caucasus region of Georgia and spread rapidly to over 80 countries across Europe (Eastern Europe, 2007; European Union, 2014), Asia (China 2018), the Pacific (Oceania 2019), and the Caribbean (Dominican Republic, Haiti, 2021; https://www.woah.org/en/disease/african-swine-fever), becoming a major threat to the global pig population and global food security.
ASF has a range of clinical signs, from peracute to subclinical.2,7 The primary viral detection method used in animals with acute or subacute ASFV infections is PCR. Serologic detection of ASFV antibody should be included, particularly in animals with chronic or subclinical infections or in recovered animals in which only antibodies but not virus are present.1,5 Several ELISAs are commercially available for detecting antibodies against ASFV. The World Organisation for Animal Health (WOAH) recommends that ELISA-positive samples should be confirmed by a second serologic method, such as an indirect immunofluorescent assay, indirect immunoperoxidase test (IPT), or immunoblot test. 10 IPT is one of the most common methods used as a confirmatory test given its high specificity and sensitivity; however, no commercial IPT kit is available. 4
We obtained the IPT antigen plate and the reference ASFV antiserum panel from the European Union Reference Laboratory for African Swine Fever (EURL-ASF) at the Centro De Investigación en Sanidad Animal–Instituto Nacional de Investigaciones Agrarias (CISA-INIA; Madrid, Spain). As a WOAH ASF reference laboratory, the USDA Foreign Animal Disease Diagnostic Laboratory (FADDL; Plum Island, NY, USA) needs to establish an IPT for in-house confirmatory testing and to support other WOAH member countries in North America for ASFV antibody testing. WOAH has outlined the procedure for preparing ASFV antigen plates for IPT with Vero or MS cells. 10 One of the virus strains commonly used for infecting the cells on the antigen plate is BA71V, 4 which is a highly attenuated and Vero cell–adapted genotype I ASFV strain with high tissue culture passages. Its genome is ~20 kb shorter than the Georgia 2007 strain. The impact of the mutation on the ASFV antigen profile is not clear. ASFV strain Georgia 2007/1 is a prototype of the currently circulating genotype II ASFV. The Georgia strain has been adapted on the MA-104 cell line (ATCC CRL-2378.1) with a lower tissue culture passage (25 passages). The adapted strain preserved most biologic and pathologic characteristics of the wild-type strain.
Before preparing the ASFV antigen plate, we optimized several parameters, such as the seeding density of MA-104 cells, multiplicity of infection (MOI), duration of infection, and fixation method. Briefly, a 96-well microplate (Stripwell 9102; Costar) was seeded with 100 µL/well of an actively growing MA-104 cell suspension at a concentration of 1.5 × 105 cells/mL in Dulbecco modified Eagle medium (DMEM 12-604F; Lonza) supplemented with 10% fetal bovine serum (FBS 12107C; MilliporeSigma) and incubated at 37°C in a 5% CO2 atmosphere for 24 h. The monolayer at 80–90% confluence was infected with the ASFV Georgia 2007/1 strain at 0.025–0.05 MOI and incubated at 37°C in a 5% CO2 atmosphere for 24 h. The infected monolayers were fixed with an acetone:methanol (30:70%) mixture (stored at 4–8°C) for 8 min at room temperature. Finally, the plates were air-dried, sealed, and stored at –70°C for use.
We performed our in-house IPT following the WOAH procedure with modifications. 10 Briefly, the IPT plate was blocked with 120 µL/well of blocking buffer [PBS, pH 7.2 with 0.05% Tween 20 (P9416; MilliporeSigma), 2.1% NaCl, and 10% KPL milk diluent/blocking concentrate kit (5140-0011; SeraCare)] at 37°C for 60 min. After blocking, 100 μL/well test sample, negative control, or positive control (mixing 25 μL of sample or control with 975 μL of blocking buffer, 1:40 dilution) were added to wells in duplicate and incubated at 37°C for 45 min. Recombinant protein A peroxidase conjugate (Pierce 32400, Invitrogen) was used at a 1:2,000 dilution in blocking buffer. The substrate, 3-amino-9-ethylcarbazole (ImmPACT AEC SK-4205; Vector), was freshly prepared according to the manufacturer’s instruction, and 50 µL was added to each well and incubated at room temperature for 8 min or until the negative control began to color. The results were read with an inverted optical microscope. The intense red cytoplasmic precipitation was interpreted as positive for ASFV antibody; the absence of red precipitation was interpreted as negative for ASFV antibodies.
The analytic sensitivity of the in-house IPT was evaluated by direct comparison between the in-house and reference IPT (obtained from EURL-ASF–CISA-INIA) using 9 ASFV antibody–positive serum samples (8 reference antibody–positive samples were obtained from EURL-ASF–CISA-INIA; 1 hyperimmune serum was from FADDL). The samples were 2-fold serially diluted to determine the endpoint dilution in which IPT was still positive for ASFV antibody. The results indicated that the endpoint dilutions in our in-house IPT were either equal to that of the reference IPT (4 samples) or 1 (4 samples) or 2 (1 sample) dilutions higher than that of the reference IPT (Fig. 1).
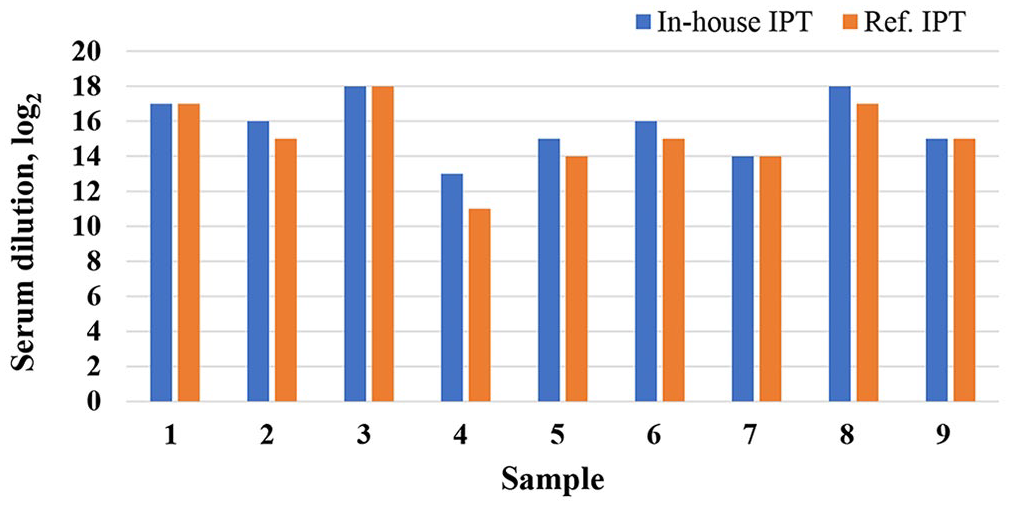
Assessment of the comparative diagnostic sensitivity between in-house and reference indirect immunoperoxidase tests (IPTs). We used 9 African swine fever virus (ASFV) reference sera; 1 sample (sample 1) was produced at the USDA Foreign Animal Disease Diagnostic Laboratory (Plum Island, NY, USA), and 8 sera (samples 2–9) were obtained from the European Union Reference Laboratory for African Swine Fever (Madrid, Spain). The sera were 2-fold diluted in blocking buffer, and the endpoint dilutions were determined for both IPTs. The reciprocal of the endpoint dilution (log2) of our in-house IPT (blue bars) was compared with that of the reference IPT (orange bars). The endpoint dilution of our in-house IPT was either equal to that of the reference IPT (4 samples), or 1 (4 samples) or 2 (1 sample) dilutions higher than that of the reference IPT.
The analytic specificity of the in-house IPT was evaluated by using 139 swine sera collected from domestic naïve pigs (100 samples were received from the classical swine fever virus [CSFV] surveillance program, and 39 were from experimental pigs collected at 0 dpi at FADDL). All samples were confirmed antibody-negative against ASFV by the reference IPT; the results of our in-house IPT were also negative. The analytic specificity of the in-house IPT was 100% (139 of 139) with 95% CI of 97–100%, estimated by the Agresti–Coull method.
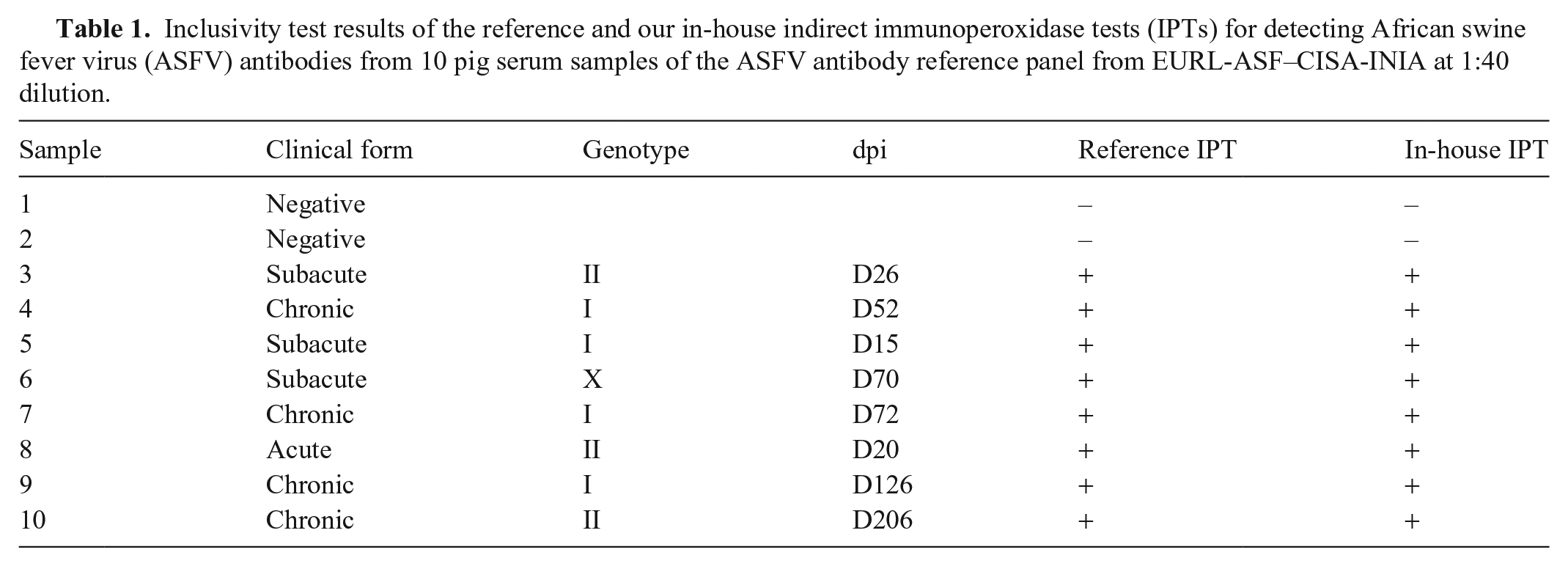
Our in-house IPT must be able to detect the specific antibody in serum samples collected from animals at different clinical stages. We evaluated the inclusivity of our in-house IPT using an ASFV antibody reference panel obtained from EURL-ASF–CISA-INIA, which includes 10 pig serum samples covering several ASFV genotypes and different clinical presentations. The samples were diluted at 1:40 in blocking buffer and tested with our in-house IPT. Our in-house IPT identified all 10 samples correctly (Table 1), suggesting that the inclusivity of the in-house IPT was 100% (10 of 10) with 95% CI of 66–100% estimated by the Wilson score with continuity correction.
Inclusivity test results of the reference and our in-house indirect immunoperoxidase tests (IPTs) for detecting African swine fever virus (ASFV) antibodies from 10 pig serum samples of the ASFV antibody reference panel from EURL-ASF–CISA-INIA at 1:40 dilution.
The diagnostic sensitivity and specificity of the in-house IPT was evaluated by using 42 ASFV antibody–positive sera collected from pigs experimentally infected with ASFV and 62 ASFV antibody–negative pig serum samples. These ASFV antibody–negative samples that were pre-tested using the reference ASFV IPT including 13 CSFV, 18 foot-and-mouth disease virus, 14 vesicular exanthema of swine virus, 1 porcine epidemic diarrhea virus, 2 swine influenza A virus, 7 porcine reproductive and respiratory syndrome virus, 1 swine pox virus, 1 senecavirus A, and 5 porcine circovirus 2 positive serum samples. The positive and negative samples were also tested using a commercial blocking ELISA (Ingezim PPA Compac, 11.PPA.K3; Gold Standard Diagnostics) validated and routinely used for detecting ASFV antibody at FADDL. The 42 ASFV antibody–positive samples tested positive, and the 62 ASFV antibody–negative samples tested negative using our in-house IPT. The estimated diagnostic sensitivity and specificity of the in-house IPT were 100% with 95% CI of 90–100% and 93–100%, respectively. Our in-house IPT did not cross-react to major transboundary and common domestic pig viral agents. In comparison, 40 of 42 ASFV antibody–positive samples tested positive and 2 were negative using the blocking ELISA, suggesting a sensitivity of 95.2% (40 of 42) with a 95% CI of 84–99%. The 62 ASFV antibody–negative samples also tested negative with the blocking ELISA, suggesting a specificity of 100% (62 of 62) with a 95% CI of 93–100% (Table 2).
The diagnostic sensitivity and specificity of the in-house indirect immunoperoxidase test (IPT) and the blocking ELISA compared with the reference IPT using serum samples with African swine fever virus antibody status confirmed by the reference IPT at a 1:40 dilution.

We further evaluated the diagnostic sensitivity and diagnostic specificity of our in-house IPT with 101 field samples collected by FADDL from recent ASF outbreaks in the Caribbean region. These samples were diluted at 1:40 and tested using the reference IPT, our in-house IPT, and the commercial blocking ELISA. We found that 49 of 101 samples (48.5%) were antibody-positive against ASFV using the reference IPT and our in-house IPT, and 48 of 101 samples (47.5%) were antibody-positive against ASFV using the blocking ELISA. Additionally, 52 of 101 samples (51.5%) were antibody-negative against ASFV using the reference IPT and our in-house IPT, and 53 of 101 samples (52.5%) were antibody-negative against ASFV using the commercial blocking ELISA (Table 3). Using the results of the EURL-ASF–CISA-INIA IPT as the reference, the diagnostic sensitivity and specificity of our in-house IPT and the blocking ELISA were estimated. The comparative diagnostic sensitivity and specificity of our in-house IPT were 100% (49 of 49) and 100% (52 of 52), respectively; type I false-positive and type II false-negative errors were not identified. The comparative sensitivity of the blocking ELISA was 89.8% (44 of 49), and the comparative specificity was 92.3% (48 of 52). The blocking ELISA had 7.7% (4 of 52) type I and 10.2% (5 of 49) type II errors. These results aligned with previous reports. 4
The diagnostic sensitivity and specificity of the in-house indirect immunoperoxidase test (IPT) and the blocking ELISA compared with the reference IPT with unknown status samples at 1:40 dilution.

ELISA and IPT are 2 antibody detection methods with distinct methodologies, each with advantages and disadvantages. ELISA allows rapid testing of large numbers of samples and can be automated readily. As a primary screening method, low percentages of type I errors with ELISA are expected and can be corrected during the confirmatory test with IPT. Although IPT may not be able to handle screening a large number of samples because it is a labor-intensive method and requires specific training to perform the test, it is an ideal method for a confirmatory test given its greater specificity and sensitivity.
Footnotes
Acknowledgements
We thank Heather Petrowski and Dr. Robin Holland in the Diagnostic Services Section of FADDL for providing reference materials. We thank Drs. Amanda Kortum and Dillon McBride for proofreading the manuscript. We also thank Dr. Gleeson Murphy for reviewing the manuscript and sharing his pearls of wisdom with us. His questions and comments improved the quality of the manuscript.
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
Our study was supported by the general fund of the NVSL. The findings and conclusions in this publication are those of the authors and should not be construed to represent any official USDA or U.S. government determination or policy.