
Abstract
Rapid growth in aquaculture has resulted in high-density production systems in ecologically and geographically novel conditions in which the emergence of diseases is inevitable. Well-characterized methods for detection and surveillance of infectious diseases are vital for rapid identification, response, and recovery to protect economic and food security. We implemented a proof-of-concept approach for virus detection using a known high-consequence fish pathogen, infectious salmon anemia virus (ISAV), as the archetypal pathogen. In fish infected with ISAV, we integrated histopathology, virus isolation, whole-genome sequencing (WGS), electron microscopy (EM), in situ hybridization (ISH), and reverse transcription real-time PCR (RT-rtPCR). Fresh-frozen and formalin-fixed tissues were collected from virus-infected, control, and sham-infected Atlantic salmon (Salmo salar). Microscopic differences were not evident between uninfected and infected fish. Viral cytopathic effect was observed in cell cultures inoculated with fresh-frozen tissue homogenates from 3 of 3 ISAV-infected and 0 of 4 uninfected or sham-infected fish. The ISAV genome was detected by shotgun metagenomics in RNA extracted from the medium from 3 of 3 inoculated cell cultures, 3 of 3 infected fish, and 0 of 4 uninfected or sham-infected fish, yielding sufficient coverage for de novo assembly. An ISH probe against ISAV revealed ISAV genome in multiple organs, with abundance in renal hematopoietic tissue. Virus was detected by RT-rtPCR in gill, heart, kidney, liver, and spleen. EM and metagenomic WGS from tissues were challenging and unsuccessful. Our proof-of-concept methodology has promise for detection and characterization of unknown aquatic pathogens and also highlights some associated methodology challenges that require additional investigation.
As consumer demand for fish and other aquatic species continues to rise, ecologic pressures change, and aquaculture practices evolve, the emergence of novel and uncharacterized viral diseases that threaten the industry is inevitable. Measures to safeguard the health of aquaculture species are a major priority of the U.S. Department of Agriculture (USDA), one aspect of which focuses on the rapid detection and response to emerging disease threats per the 2021–2023 National Aquaculture Health Plan and Standards. 39 To meet these anticipated needs, it is vital to prepare veterinary diagnostic laboratories with the equipment, personnel, and training to detect such threats.
Scientists find themselves in a virus discovery boom time given advances in molecular techniques that have greatly improved pathogen detection and characterization resulting in a significant increase in the number of novel and newly described viruses. 14 Traditional methods of virus discovery include microscopic examination of infected tissues for viral-induced tissue changes (such as viral inclusion bodies), virus isolation (VI) in cell culture systems, electron microscopy (EM), immunohistochemistry (IHC), in situ hybridization (ISH), and molecular detection of segments of viral genomes by PCR assays and sequencing. There are shortcomings with each of these methods, particularly for viruses that do not result in characteristic histologic lesions, are challenging to grow in cell culture, or are uncharacterized; thus, targeted antigenic and genomic assays fail.
Next-generation sequencing (NGS) has transformed the field of microbial detection and characterization; however, many gaps still exist. For one, many commercial platforms and molecular methods are available, which is beneficial for optimizing characterization, but standardization is lacking between veterinary investigatory laboratories. In a recent survey of U.S. laboratories within the National Animal Health Laboratory Network (NAHLN), it was found that many different sequence platforms were being used, and some laboratories had more than one platform available. 23 Application of NGS-based protocols for nucleic acids (NAs) extracted directly from various sample types has been tested in various diagnostic laboratories, although no standard technique exists.29,43
Metagenomic sequencing enables unbiased pathogen detection with rapid runtimes and real-time data analysis on some platforms, which reduces the need for ordering multiple molecular assays on a single sample when screening for more than one agent. Data analysis for known agents is relatively straightforward, with metagenomic sequence-capture offering the ability to screen for multiple variants without relying on species-wide or strain-specific primers that are used in conventional and real-time PCR (rtPCR) assays. However, these targeted strategies rely on the existence of well-characterized pathogen genomes.
Another major challenge in pathogen discovery is attributing or associating disease causality to a particular microorganism detected by NGS modalities, given that the commensal microbiomes of healthy individuals are not characterized for most aquatic animal hosts. Fulfillment of Koch postulates becomes problematic with viruses, especially when they are not culturable, and intensive investigatory studies of causality are often not feasible when a timely response is needed in outbreak situations. 38 Given the abundant data output from NGS platforms, especially when analyzing samples by metagenomics, it becomes critical to at least associate virus or viral components with lesions in infected tissue to attribute morbidity or mortality to the detected organism. Advancements in ISH techniques such as RNAscope have improved the ease of visualizing viral genome in formalin-fixed, paraffin-embedded tissues and have been used widely to initiate suggestions of disease causality.19,31,36,41
Our objective was to develop and demonstrate competency of an integrated pipeline for the detection and characterization of aquatic viruses at the Washington Animal Disease Diagnostic Laboratory (WADDL; Washington State University, Pullman, WA, USA) using salmon experimentally infected with a well-known, transboundary WOAH-reportable disease agent, infectious salmon anemia virus (ISAV; Orthomyxoviridae, Isavirus salaris). Our model is based on using various assays that exist in the laboratory system (such as histopathology, VI, and EM), acquiring competency, and integrating NGS technologies and ISH to improve the overall diagnostic system. Our major aim was to develop a method for using third-generation sequencing and open-source, widely available bioinformatics tools to characterize ISAV in order to standardize the approach of sample preparation and data analysis for applicability to the discovery of novel viruses and rapid detection of emerging infectious diseases. The diagnostic pipeline described in our project (Fig. 1) may serve as a model for other veterinary diagnostic laboratories and may increase the preparedness of the NAHLN to detect and respond quickly to emerging threats to aquaculture or other agriculture systems.
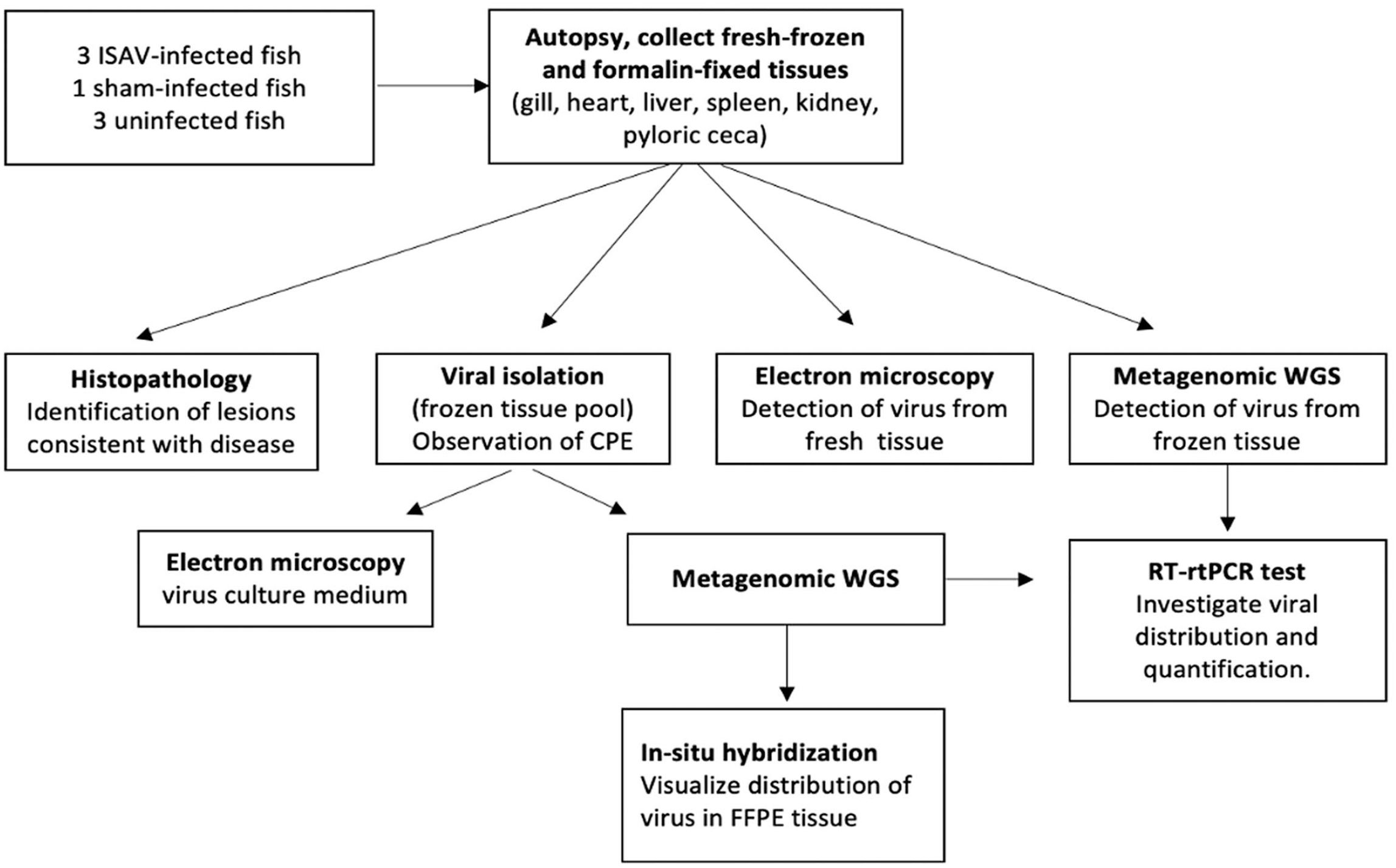
Experimental pipeline for detection and characterization of infectious salmon anemia virus (ISAV) in fish.
Materials and methods
Animal infection and sample collection
Fish were housed at the National Veterinary Services Laboratories (NVSL; Ames, IA, USA), and experimental procedures were approved by the NVSL/Center for Veterinary Biologics Animal Care and Use Committee. Twenty-three 1-y-old Atlantic salmon (Salmo salar) parr averaging 58.4 g and 17 cm in length were separated into 3 experimental groups: experimentally infected (9 fish), control (9 fish), and sham-infected (5 fish). On day 0, fish in the infected group each received a 100-µL intracoelomic injection containing ~1.0 × 105.3 TCID50 of highly polymorphic region (HPR)-deleted North American ISAV. Sham-infected fish each received a 100-µL intracoelomic injection of 0.01 M PBS. On 7, 9, and 11 d post-injection (dpi), fish from each group were euthanized by immersion in a buffered solution of 250 mg/L tricaine methanesulfonate (Tricaine-S; Syndel). These collection times were chosen based on previous work demonstrating peak viremia at 11 dpi, 1 thus ensuring that tissues would be infected. Samples of gill, liver, spleen, heart, and kidney were collected from each fish, split into duplicates with one set placed in 10% neutral-buffered formalin, and the other into sterile bags and immediately placed on dry ice (i.e., fresh-frozen tissue). Formalin samples were shipped from NVSL to WADDL at room temperature; fresh-frozen tissues were sent on dry ice. Frozen tissues were then held in −80°C freezers until processed for testing. Only the tissues that were collected at 11 dpi (3 ISAV experimentally infected, 3 control, and 1 sham-infected fish) were used for our investigation; remaining tissues were held for other research not presented here.
PCR confirmation of infected fish
Gill clips were collected from fish on each collection day for testing by reverse-transcription rtPCR (RT-rtPCR) to ensure that experimental infection had resulted in systemic viral replication prior to shipping samples to WADDL. Testing followed the USDA-established ISAV testing workflow. 2 Briefly, gill tissues were homogenized by microbead beating, and total NA was extracted from gill tissue homogenate supernatants (MagMAX-96 viral RNA isolation kit; Life Technologies). Eluted RNA was evaluated for ISAV using the WOAH-recommended segment 8 TaqMan probe–based detection method that was modified to a one-step RT-rtPCR multiplex assay.
Histopathology
Formalin-fixed tissues were trimmed, processed, and embedded in paraffin wax within 24 h of receiving them at WADDL. H&E-stained glass slides were prepared from all fish, and included gills, heart, kidney, intestines, pancreas, liver, and spleen. Two American College of Veterinary Pathologists board-certified pathologists (L.B. Williams, C.D. Eckstrand) reviewed anonymously all slides for histopathologic changes.
Viral culture
Tissue pools containing gill filament, heart, kidney, liver, and spleen from all treatment groups collected at 11 dpi were homogenized following methods in the Manual of Diagnostic Tests for Aquatic Animals, 42 and homogenate supernatants were used for VI. Tissue homogenates were processed at a 1:1 ratio with calcium-magnesium–free PBS (CMF-PBS; Fisher Scientific) and centrifuged at 2,000 × g at 4°C for 15 min. For each sample, 200 µL of supernatant was collected and added to 800 µL of processing medium (RPMI [Corning]–5% FBS [Hyclone; Cytiva] + 1% penicillin–streptomycin [PenStrep] + 0.1% gentamicin + 0.5% amphotericin B [Fungizone; Sigma-Aldrich]) for a dilution of 1:10. Two serial 10-fold dilutions (1:100; 1:1,000) of the 1:10 sample were prepared. Medium was removed from 24-well plates previously seeded with salmon head kidney (SHK) cells at 80–90% confluency, and 100 µL of each prepared dilution was inoculated into 1 well for each sample. On each plate, 2 negative control wells were inoculated with 100 µL of processing medium only. Inoculated plates were placed into sealable bags, positioned on a plate rocker, and incubated at 15°C for 3–4 h. At the end of incubation, 1.0 mL of maintenance feed medium (L-15 [+ 2-mercaptoethanol; Sigma-Aldrich] + 5% FBS + 0.5% penicillin–streptomycin + 2% L-glutamine [Fisher Scientific] + 0.1% gentamicin + 0.25% amphotericin B) was added to each well. The plates were incubated at 15°C for 14 d. Each well was observed for cytotoxic effect (CTE), microbial contamination, and cytopathic effect (CPE) 3 times per week. If signs of CTE or CPE (such as rounding of cells, intercellular gaps, cell death) were observed, 0.5 mL of maintenance feed medium was added to the culture to determine CTE or CPE. Primary cell cultures at 14 d post-inoculation were sub-passaged by harvesting all medium and cells, centrifuging for 15 min at 2,000 × g at 4°C, and inoculating 100 µL of the supernatant in duplicate onto newly plated cells. The plates were again incubated at 15°C for 14 d and observed 3 times per week. Two of the infected samples began showing CPE at day 12 on second passage; therefore, all samples were harvested and inoculated onto a third passage at day 16. Plates continued to be observed 3 times per week. When CPE was observed on passage 3, culture fluid and cells were harvested into cryovials and transferred to –80°C freezers for longer-term storage. Positive control wells were inoculated separately with ISAV-positive controls to ensure test cell line susceptibility.
Electron microscopy
Formvar-carbon–coated transmission EM (TEM) grids (Electron Microscopy Services) were hydrophilically polarized using the “air positive negative surface” clean air setting on the GloQube glow discharge apparatus (Quorum Technologies). Cell culture supernatants collected from culture isolates showing CPE were applied to grids using the drop-by-drop method 15 or by precipitant deposition onto the grid surface using an air-driven ultracentrifuge (Airfuge; Beckman Coulter) as described previously. 8 For each mounting technique, grids were prepared in duplicate and negative-stained using 2% phosphotungstic acid or 0.5% uranyl acetate. Prepared TEM grids were visualized (FEI Technai G2 20 twin transmission electron microscope, 200 KV LaB6 source emitter, Eagle FP 5271/82 4K HR200KV digital camera; Field Emission Instruments).
Whole-genome sequencing and bioinformatics
For all VI cultures, regardless of CPE status, total NA (DNA and RNA) was extracted from whole-cell culture (UltraSens virus kit; Qiagen). Double-stranded DNAs were prepared from total RNA using a sequence-independent single-primer amplification strategy, 12 which employs random priming of a tagged random octamer sequence in the initial cDNA synthesis followed by second-strand cDNA synthesis and amplification using the tagged primer without the random octamer. Amplified cDNAs were used as input for Nanopore library construction on the ligation sequencing kit (SQK-LSK109) and sequenced for 72 h with no reloading, with each sample on an individual R9.4.1 flow cell (FLO-MIN106) on the GridION platform (Nanopore Technologies). The run was initiated using MinKnow v.20.10.6 installed on the ONT GridION release configuration 4.1.15 custom OS built on Ubuntu 16.04. Raw data were base-called on the GridION GPU at the time of sequencing in high-accuracy mode using Guppy v.4.2.3. For low-depth short reads later used to polish the draft genome, these same cDNAs were prepared using the Illumina DNA Prep library kit with Nextera indices and sequenced on a partial lane of the MiSeq v3 reagent kit (600-cycle; Illumina). Illumina data were demultiplexed and base-called on BaseSpace (https://basespace.illumina.com/) using FastQ Generation v.1.0.0.
Given that the final application of our pipeline is intended for suspected viruses, data were analyzed as though the cultured agent was unknown. After all sequencing was complete, data were aligned to viral reference sequences from NCBI (n = 13,580) 35 as a guide to cluster NA sequences by sequence identity to known viruses and reduce host reads. Next, raw data were run through Diamond 9 BLASTX for high-throughput analysis of protein-coding sequences matched against the Reference Viral Protein Database (RVDB v.21.0). 20 Clustered viral NA and protein sequences were used to identify signatures of likely viral candidates that may infect aquatic vertebrates. In the case of our known agent sample, NA and protein identities both strongly suggested the presence of ISAV in terms of sequence concordance and genome coverage. All candidate viral reads identified by nucleotide and protein homology, or those classified by ONT’s metagenomic classification workflow What’s in My Pot (WIMP) 24 on the Epi2Me webserver (https://epi2me.nanoporetech.com) during real-time analysis, including cyprinid herpesvirus 1 and 2 and fragments of several retroviruses, were manually investigated by re-mapping all raw reads to each suggested pathogen individually and evaluating mapping quality and query coverage. In cases of a questionable alignment or low-quality reference hit, raw reads were submitted to NCBI Megablast against the nr/nt database (https://blast.ncbi.nlm.nih.gov/Blast.cgi). By this method of re-mapping and BLAST searching, whole raw reads classified as herpesviruses and retroviruses produced high-scoring matches to putative elements of various fish genomes, and thus were excluded from viral candidacy.
To evaluate ISAV as a candidate, we mapped all raw reads to the concatenated reference genome for salmon isavirus (NC_06497.1, NC_006498.1, NC_006499.1, NC_006500.2, NC_006501.1, NC_006502.1, NC_006503.1, NC_006505.1), which is comprised of segments from various ISAV isolates, including CCBB and 810/9/00.13,26 To test the ability of Nanopore data to generate whole-genome assemblies of short-segmented RNA viruses, ISAV-mapped GridION reads were de novo assembled in a long-read first paradigm using Canu v.2.1. 27 Similarly, MiSeq reads were mapped to ISAV and de novo assembled using SPAdes v.3.15. 4 Finally, GridION and MiSeq reads were assembled in a hybrid strategy using SPAdes for contig extension only, Canu and hybrid SPAdes contigs were aligned in Geneious Prime v.2021.1.1, 6 and consensus sequences were concatenated into an initial draft genome. The draft genome was polished with raw reads using Racon 40 and Medaka. 37 Single-nucleotide variations (SNVs) in the draft genome were considered accurate if the position had sufficient depth of GridION or MiSeq reads (100× and 50×, respectively) and if the base was called the same by both technologies.
RT-rtPCR of fresh-frozen tissues and virus cultures
Previously described primer sequence targets were present in the whole-genome sequencing (WGS) draft genome. Individual fresh-frozen gill, liver, kidney, and spleen tissues from each day-11 fish were placed in a bead-beating tube with silica beads and cell culture medium, homogenized in a 10% w/v solution, and extracted as described previously. 22 As well, RNA extraction and RT-rtPCR were performed in duplicate from 1 of 2 inoculated cell culture supernatants for each day-11 fish using the same described RT-rtPCR protocol. 22 ISAV was considered detected by RT-rtPCR if there was a Ct value before 40 cycles and considered not detected if after 40 cycles.
For tissue sequencing, tissue pool homogenates used to seed VI cultures (50% w/v in CMF-PBS) were diluted to a 10% w/v suspension by the addition of stock DMEM (Invitrogen). Diluted homogenates were vortexed and pelleted (5 min × 3,500 rcf at room temperature), and 500 µL of cleared supernatant was extracted (UltraSens kit; Qiagen) according to the manufacturer’s instructions. Double-stranded cDNAs were synthesized from total NA, and Nanopore libraries were constructed as above. A second attempt to detect ISAV in infected tissues utilized kidney homogenates from 2 ISAV-infected fish.
ISH for tissue localization of viral genome
ISH utilized RNAscope (Advanced Cell Diagnostics [ACD]) technology to visualize the presence and location of viral RNA in tissues harvested from infected fish. A set of anti-sense ISAV-specific RNA probes, comprised of 20 Z pairs targeting nucleotides 5–976 of segment 2 containing the viral polymerase of the viral genome (GenBank AF_262399.1), was developed by ACD and performed as described previously.16,17 The assembled WGS draft genome was a 99% match to this ISH probe–targeted region. This assay was performed according to the manufacturer’s protocols for RNAscope 2.5 HD Red detection kit (ACD) with the following specific conditions. Paraffin-embedded tissues from uninoculated, sham-infected, and ISAV-infected fish harvested at 11 dpi were sectioned at 4 µm on positively charged glass slides. Samples were slowly submerged in lightly boiling target retrieval solution (ACD) for 15 min, followed by application and incubation of Protease Plus (ACD) at 40°C for 20 min. In addition, a positive control probe targeting nucleotides 20–934 of a constitutively expressed Atlantic salmon gene peptidylprolyl isomerase B (ppib, GenBank NM_001140870.2) and a negative control probe for the dihydrodipicolinate reductase gene (dapB) of Bacillus subtilis strain SMY (GenBank EF_191515) were applied to test tissues.
Results
Virus culture
CPE characterized by dark granular aggregates of cells surrounded by individual elongated cells was observed in 3 of 3 ISAV-infected fish harvested at 11 dpi after passage 2 or 3 (starting at day 12 post-culture) and in none of the sham-infected or uninfected fish.
Histopathology
By light microscopy, the 3 ISAV-uninfected fish had minimal histologic changes in the gills, heart, liver, spleen, kidney, pyloric ceca, and pancreas, including rare individualized apoptotic epithelial cells in the pyloric ceca (2 of 3) and a few lymphocytes within secondary gill lamellae (1 of 3). These findings were considered clinically insignificant. No microscopic changes were appreciated in the one sham-infected fish. In the 3 ISAV-infected fish, there was mild lymphohistiocytic epicardial steatitis (1 of 3), very rare hypercellularity of secondary gill lamellae (1 of 3), and splenic congestion (2 of 3). Overall, there were no histologically consistent abnormalities among ISAV-infected fish or between ISAV-infected and uninfected or sham-infected fish.
Electron microscopy
Negative staining of culture medium in which CPE was observed revealed viral particles that were pleomorphic (Fig. 2). Viral particles were ~100 nm diameter, with pleomorphism observed in the viral envelope.
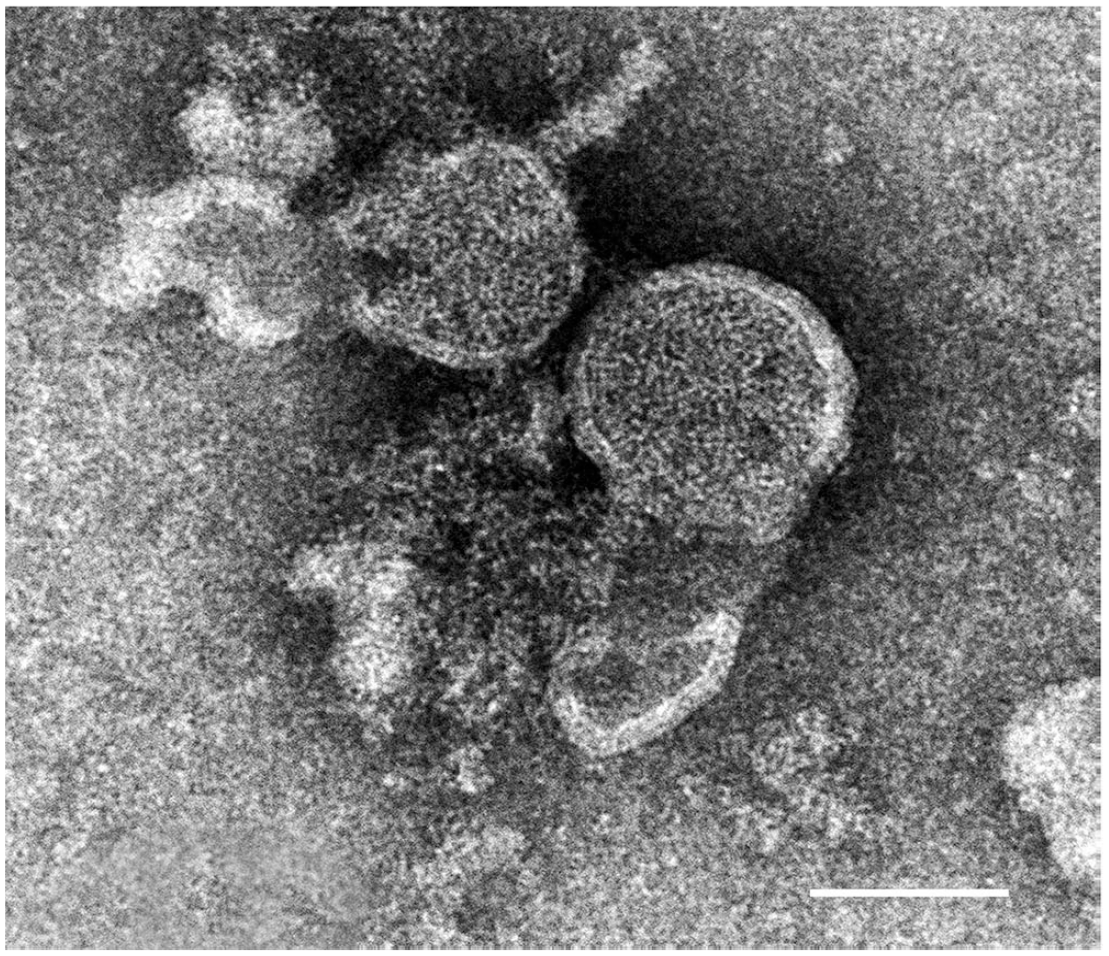
Electron microscopic image of infectious salmon anemia virus (ISAV) viral particle from negatively stained virus culture medium of a day-11 ISAV-infected fish. Bar = 100 nm.
Genomic sequencing of ISAV
From whole tissue, ISAV was detected at low levels in the kidney but not in pooled tissues (52 Nanopore reads/6,187,260 = 0.0008%) with 12.8 kbp aligned to the whole genome at an average depth of 3 (in non-zero regions of coverage) and the greatest breadth of coverage across segment 5 (81% of bases coverage ≥ 1). This amount of coverage and depth is not sufficient for de novo whole-genome assembly, but it is a strong signature of viral presence in this tissue.
Using only Nanopore data from CPE-positive virus culture medium extracts, the total length of our first draft genome assembly was ~96% of the reference genome length (12,190 bp). Incorporating Illumina reads with our Nanopore data in a hybrid SPAdes short-read first assembly strategy produced a higher-confidence draft genome that was shortened in length compared to the first draft because of uneven distribution of reads along the segmented genome. Contigs from both draft assemblies were aligned and combined to generate a 12,622-bp polished consensus draft genome. Using NCBI Megablast against the non-redundant nucleotide database, the draft genome sequences matched GenBank accessions KX424582.1, KX424583.1, KX424584.1, KX424586.1, KX424588.1, and KX424589.1 as the highest-scoring hit for 6 of the 8 segments, which seem to belong to a single strain designated “ISAV isolate NA-HPR4 (970-1)” (Suppl. Table 1). 30 Reference segment coverage of assembled contigs ranged from 65.0–102% because some of the assembled contigs were longer than the reference accession lengths and some shorter as a result of markedly lower coverage on both platforms across the shortest ISAV segment. To elucidate whole-genome comparison between our draft genome and publicly available whole ISAV genomes, we mapped the data to GenBank KX424582–KX424589 and found that raw reads aligned to these 8 segments with high concordance, yielding a consensus genome with ≥ 14 high-quality SNVs—5 of which confer amino acid substitutions—with GridION sequencing depth > 100× and/or MiSeq depth > 50× (Suppl. Table 2).
ISH for tissue localization of ISAV genome
Viral genome (RNA) was visualized in the gills, heart, liver, spleen, head kidney, and pyloric ceca of the day-11 ISAV-infected fish, and was not detected in any tissues of the uninfected or sham-infected fish. In all 3 ISAV-infected fish, infected cells were observed in the superficial cells of the gill, interstitium of the heart, along the sinusoids of the liver, which are presumed endothelial cells (Fig. 3), in the white pulp of the spleen, the hematopoietic interstitium of the head kidney, and the lamina propria and interstitium of the muscularis of the pyloric ceca (Fig. 4). Viral RNA was not visualized in pancreatic parenchymal cells in any of the infected fish, but there were occasional positive cells in the interstitium of the supporting peritoneal adipose tissue.
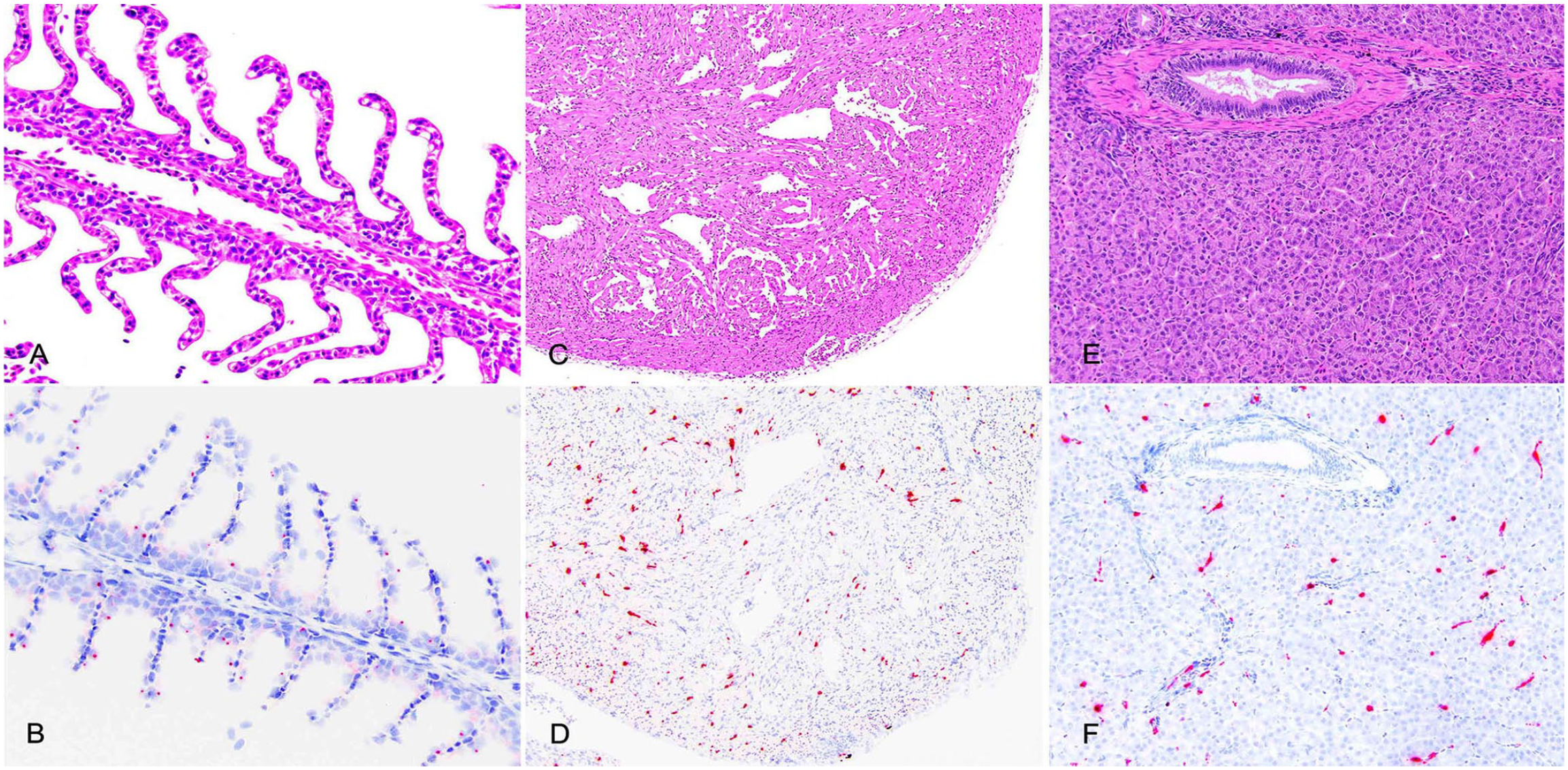
Organ histology (A, C, E; H&E) and corresponding distribution of infectious salmon anemia virus (ISAV) genome by in situ hybridization (ISH; B, D, F; hematoxylin counterstain of ISH). ISAV RNA is randomly distributed in the gill filaments (A, B), the interstitial connective tissue of the myocardium (C, D), and cells lining the sinusoids (presumed endothelial cells) of the liver (E, F).
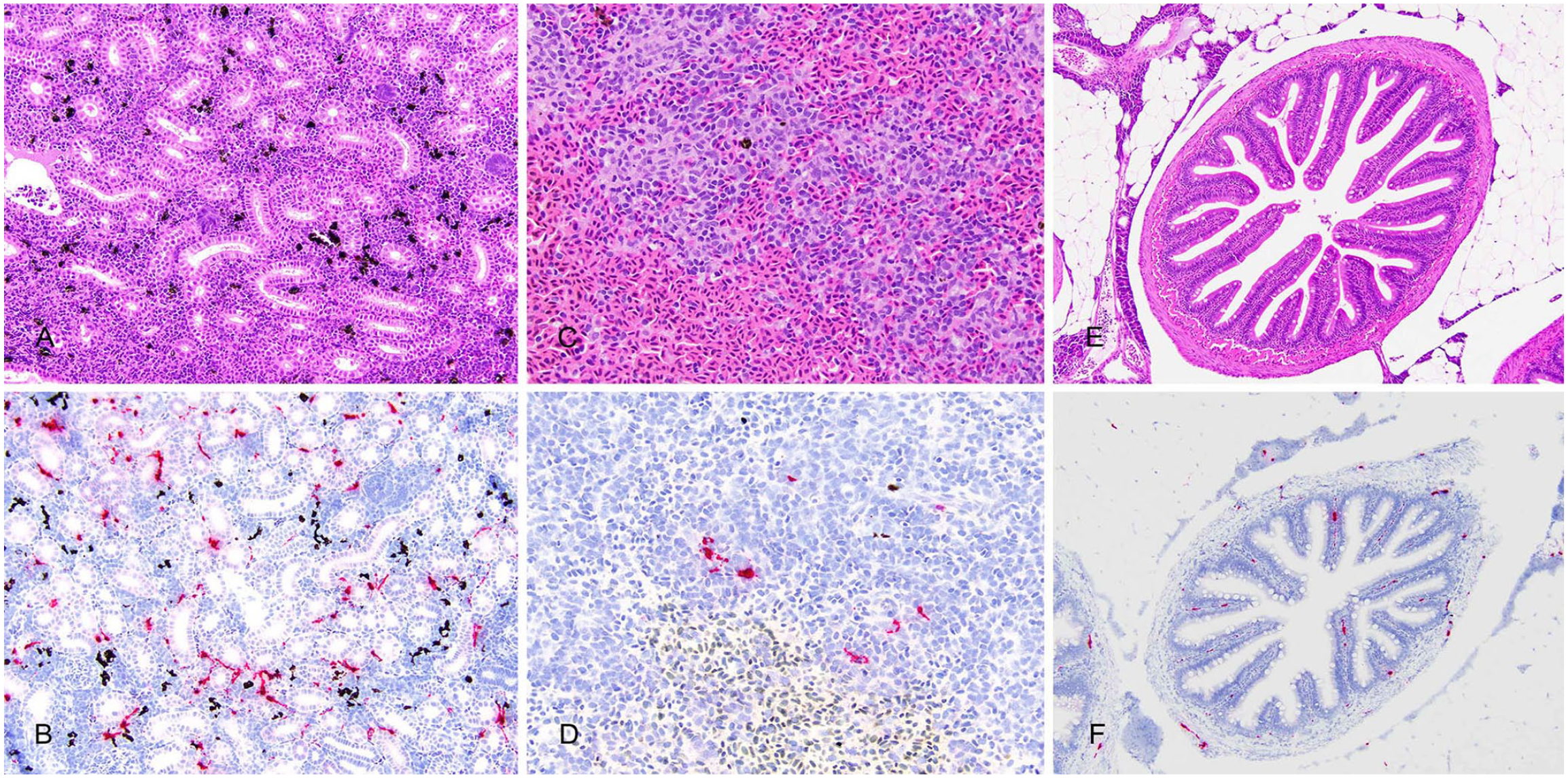
Organ histology (A, C, E; H&E) and corresponding distribution of infectious salmon anemia virus (ISAV) genome by in situ hybridization (ISH; B, D, F; hematoxylin counterstain of ISH). ISAV RNA is concentrated in the hematopoietic interstitium of the head kidney (A, B), the white pulp of the spleen (C, D), and the lamina propria and interstitial connective tissue of the intestines (E, F).
The amount of genome present by ISH was quantified in the gill, kidney, liver, and spleen by counting numbers of ISH dots per 400× magnification field over 3 fields and calculating an average (Fig. 5). Relative to other organs, the head kidney had the greatest amount of viral RNA staining (> 400 dots/hp f) in all 3 ISAV-infected fish. An intermediate amount of viral RNA was seen in the heart and liver of 1 of 3 infected fish, and the remaining organs had lower amounts of genomic staining. The positive control probe demonstrated detectable RNA in both the ISAV-infected and uninfected control tissues; the negative control probe demonstrated no positive staining in ISAV-infected and uninfected fish.
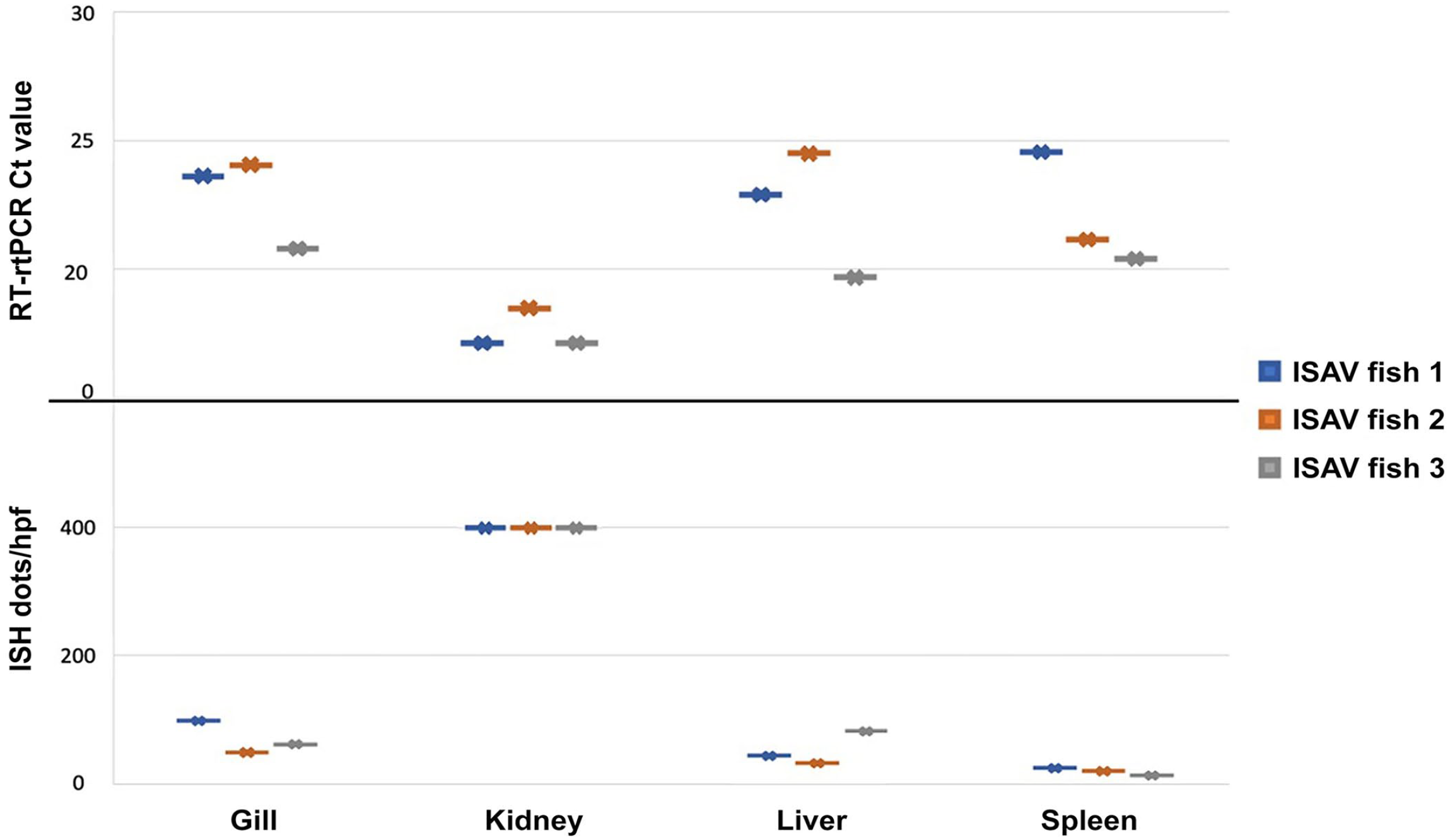
Infectious salmon anemia virus (ISAV) in tissues by reverse-transcription real-time PCR (RT-rtPCR) and in situ hybridization (ISH). The Ct values for each day-11 ISAV-infected fish was determined by RT-PCR, as well as amount of viral genome visualized in each of the corresponding tissues by ISH (represented as average dots per 400× magnification field). In both tests, there is an apparent trend that kidney tissue contains a greater amount of virus; however, this was not statistically significant when comparing means (p > 0.05).
A one-way ANOVA was performed to compare the effect of tissue type on the virus genome visualized by ISH and revealed that there was a statistical difference in Ct value of virus between at least 2 groups (F = (3, 8) = [188.406]; p = 0); however, a Tukey honestly significant difference (HSD) test for multiple comparisons indicated that the mean amount of virus per hpf was not statistically significantly different between any compared tissue types (p > 0.05).
PCR of fresh-frozen tissues and virus cultures
ISAV was detected by RT-rtPCR using primers that match target sequences identified in the WGS draft genome in gill, liver, spleen, and kidney of 3 of 3 infected fish and in no sham-infected or control fish (Fig. 5). A one-way ANOVA was performed to compare the effect of tissue type on the Ct value of virus and revealed that there was a statistical difference in Ct value of virus between at least 2 groups (F = (3, 8) = [4.2076]; p = 0.04623); however, a Tukey HSD test for multiple comparisons found that the mean Ct values were not statistically significantly different between any compared tissue types (p > 0.05). ISAV was also detected in 2 of 2 tests for 2 of the CPE-positive cell cultures and 1 of 2 tests for the third CPE-positive cell culture supernatant. ISAV was not detected in any of the CPE-negative and cell culture controls when duplicate tests were run. When comparing the trend of RT-rtPCR Ct value and the amount of virus observed by ISH, there was a clear pattern that, in both tests (RT-rtPCR and ISH), renal tissue consistently contained more virus than the other tissues examined (Fig. 5).
Discussion
Our work provides a roadmap that veterinary diagnostic laboratories could utilize for the detection and characterization of a novel virus affecting aquaculture species. We demonstrated how the integration of basic laboratory techniques of histopathology, VI, EM, and RT-rtPCR with newer NGS and ISH technologies can be used to detect and characterize a virus. These competencies utilizing sophisticated NGS and ISH technologies will help to refine our techniques for the characterization of novel and rapidly migrating viral agents in which the accumulation of mutations compromises strain detection by traditional PCR and also highlights the need for the development of new assays for emerging viruses.
We found WGS of ISAV from tissue using unbiased metagenomic sequencing to be far more challenging than WGS from propagated virus culture medium, which highlights the need for improvements in our pretreatment and enrichment techniques. In searches of Google, PubMed, and Web of Science databases, we retrieved no reports describing WGS of ISAV directly from tissue from infected fish, although partial sequencing for rapid detection and specific segment characterization is achieved more easily. 18 A rapid expansion of availability and accessibility of NGS has led to an explosion of publicly available sequences. However, sequence completeness and quality vary, most likely a direct result of the lack of standardization in viral genomic research. This is especially challenging for ISAV, given that strain names were not always consistently included in GenBank records, historical records were not updated to reflect current strain nomenclature, and complete genomes (with all 8 segments) for each strain were not always available.
Phylogenetic analyses of segments 2 and 8, encoding PB1 and 2 nonstructural proteins, respectively, support the classification of ISAV into 2 major genogroups from North America (including Canada and the USA) and Europe; however, studies have begun to classify ISAV by surface glycoprotein hemagglutinin esterase (HE, segment 6) and fusion protein (F, segment 5) that contribute to its pathogenicity.10,25,26,28 These types of targeted studies result in incomplete genomes of record for individual ISAV isolates within the NCBI database; and, to date, most attempts to describe the relatedness of different ISAV strains have been based on one or several loci, but rarely are whole, multipartite genomes considered. 25 This paucity of information combined with the high mutability of RNA viruses presents an opportunity for WGS to enhance molecular phylogenomics and outbreak tracing in aquaculture.
Similarly, the challenges we faced obtaining whole genome data from ISAV-infected tissues and purification of virus particles from infected tissue for morphologic characterization by EM proved to be difficult and time-consuming; observation of virions from viral culture medium is achieved more easily. This is not entirely surprising given the amount of host material in tissue samples; these findings suggest that EM may be best suited for samples in which virus culture is achievable and rapid detection is less urgent.
Our work strongly supports the utility of ISH in characterizing viral infections by describing the systemic distribution of the ISAV genome in infected fish tissues and its absence in uninfected fish. The visualization of viral NAs by ISH in infected, diseased animal tissues, and absence in uninfected, non-diseased tissues, supports the pathogenic significance of an organism detected from the genomic soup of data from metagenomic NGS. The utility of ISH in attributing a possible causal association between a virus and disease has been demonstrated many times by pathogen discovery groups in veterinary and human medicine.3,5,7,32,34 Although it did not occur in our case, a potential challenge of integrating ISH into a workflow may be in identifying a genomic sequence for probe design for undescribed viruses with little or no published information.
We demonstrated the distribution of ISAV genome in experimentally infected fish at 11 dpi in gills, heart, liver, spleen, intestines, and kidney. In the liver and heart, endothelial cells demonstrated positive ISAV reactivity, which supports reports that ISAV is endotheliotropic.1,21,33 Cellular localization of viral genome to superficial cells of the gill (presumed gill epithelium) has also been described with ISAV-HPR0–infected salmon. 2 To our surprise, we discovered that the hematopoietic tissue of the head kidney harbored the greatest amount of ISAV genomic material by ISH, as well as by RT-rtPCR, suggesting that further investigations may be warranted. This contrasts with previous reports that describe lesser amounts of virus concentrated in the hematopoietic tissue of the head kidney11,33 or concentrated virus in the endothelium of the glomeruli. 21 These differences may be the result of virus strain, isolate used in the study, infectious dose, days post-injection, and other unknown causes.
We have described a pipeline that was used for a culturable virus with an NCBI-curated reference genome, which improved the speed and focus of detection in our case, but which may not be the case for many viruses. The next step is to apply this virus discovery pipeline to tissues from animals experimentally infected with unculturable agents to show that shotgun metagenomics can mitigate the challenge of unbiased virus detection. Future studies should focus on efficient methods for detecting candidate viruses in infected tissue pools or investigating other less host-contaminated samples, such as blood.
A major limitation of our investigation is our attempt to compare a controlled experimental infection by a known virus with an outbreak situation of an unknown pathogen in which environmental conditions, samples, times, pathogen type, infection status, clinical signs, and many other factors may be highly variable. We attempted to mitigate testing bias by approaching all assays in an anonymized fashion; however, many outbreak factors, such as variability in samples, infection status, and clinical signs, were not accounted for. Another major challenge in pathogen discovery is the limitation in published genomic libraries for identification of novel pathogens. Thus, it is plausible that some or many of the methods outlined in our proposed pipeline may fail or falsely identify a pathogen in a real-life situation. This highlights the need to use various modalities in a disease investigation (VI, metagenomics, histopathology, EM, etc.) given that particular methods may be superior in different situations.
Supplemental Material
sj-pdf-1-vdi-10.1177_10406387231173332 – Supplemental material for Investigation of laboratory methods for characterization of aquatic viruses in fish infected experimentally with infectious salmon anemia virus
Supplemental material, sj-pdf-1-vdi-10.1177_10406387231173332 for Investigation of laboratory methods for characterization of aquatic viruses in fish infected experimentally with infectious salmon anemia virus by Chrissy D. Eckstrand, Brandi K. Torrevillas, Rebecca M. Wolking, Daniel S. Bradway, Janet V. Warg, Richard D. Clayton, Laura B. Williams, Allan P. Pessier, Joetta Lynn Reno, Kathleen M. McMenamin-Snekvik, Jim Thompson, Timothy Baszler and Kevin R. Snekvik in Journal of Veterinary Diagnostic Investigation
Supplemental Material
sj-xls-2-vdi-10.1177_10406387231173332 – Supplemental material for Investigation of laboratory methods for characterization of aquatic viruses in fish infected experimentally with infectious salmon anemia virus
Supplemental material, sj-xls-2-vdi-10.1177_10406387231173332 for Investigation of laboratory methods for characterization of aquatic viruses in fish infected experimentally with infectious salmon anemia virus by Chrissy D. Eckstrand, Brandi K. Torrevillas, Rebecca M. Wolking, Daniel S. Bradway, Janet V. Warg, Richard D. Clayton, Laura B. Williams, Allan P. Pessier, Joetta Lynn Reno, Kathleen M. McMenamin-Snekvik, Jim Thompson, Timothy Baszler and Kevin R. Snekvik in Journal of Veterinary Diagnostic Investigation
Footnotes
Acknowledgements
We acknowledge the assistance of the personnel at the Washington State University Franceschi Microscopy and Imaging Center with electron microscopy and Rachel Olson at the Washington Animal Disease Diagnostic Laboratory for assistance with MiSeq library preparation and sequencing.
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to research, authorship, and/or publication of this article.
Funding
ORCID iDs
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.