
Abstract
Rapid and reliable identification of the hemagglutinin (HA) and neuraminidase (NA) genetic clades of an influenza A virus (IAV) sequence from swine can inform control measures and multivalent vaccine composition. Current approaches to genetically characterize HA or NA sequences are based on nucleotide similarity or phylogenetic analyses. Public databases exist to acquire IAV genetic sequences for comparison, but personnel at the diagnostic or production level have difficulty in adequately updating and maintaining relevant sequence datasets for IAV in swine. Further, phylogenetic analyses are time intensive, and inference drawn from these methods is impacted by input sequence data and associated metadata. We describe here the use of the IAV multisequence identity tool as an integrated public webpage located on the Iowa State University Veterinary Diagnostic Laboratory (ISU-VDL) FLUture website: https://influenza.cvm.iastate.edu/. The multisequence identity tool uses sequence data derived from IAV-positive cases sequenced at the ISU-VDL, employs a BLAST algorithm that identifies sequences that are genetically similar to submitted query sequences, and presents a tabulation and visualization of the most genetically similar IAV sequence and associated metadata from the FLUture database. Our tool removes bioinformatic barriers and allows clients, veterinarians, and researchers to rapidly classify and identify IAV sequences similar to their own sequences to augment interpretation of results.
Influenza A virus (IAV; Alphainfluenzavirus influenzae) causes respiratory disease with elevated production costs in commercial swine production systems.5,15 Clinical signs in swine may include coughing, fever, lethargy, and a reduction of appetite that can lead to weight loss. Further production losses may be incurred through lung lesions induced by IAV that predisposes swine to secondary bacterial infections, resulting in treatment costs and increased risk of mortality.6,16 IAV outbreaks have high morbidity associated with clinical disease; thus, prevention and control are necessary to minimize animal suffering, mitigate production losses, and protect public health. Vaccination is the primary method of on-farm control of IAV endemic infections and clinical disease. However, vaccines are most effective when vaccine antigens are well-matched against circulating strains.13,14 Therefore, understanding the genetic diversity of IAV circulating within a production system is critical for making the most effective vaccine decisions.
The Iowa State University Veterinary Diagnostic Laboratory (ISU-VDL; Ames, IA, USA) processes >1,500 diagnostic swine submissions each year and sequences a subset of IAV-positive accessions for hemagglutinin (HA) and neuraminidase (NA) genes. Currently the ISU-VDL hosts >13,000 IAV sequences collected from 2003 to present, derived from 32 states across the United States. In 2018, we introduced the near-real-time epidemiology system and database, ISU FLUture, to synthesize and graphically present spatial and temporal trends in IAV sequence diversity in swine, 17 to address the need for comparing sequences of IAV field strains with genetically similar strains processed at the ISU-VDL. Our tool rapidly identifies genetically similar IAV sequences that are stored in the ISU-VDL database, including the associated case metadata, using one or multiple sequence input queries provided by users.
The ISU FLUture multisequence identity tool uses BLAST to detect and return up to 10 IAV sequences in the ISU-VDL database with ≥96% nucleotide or amino acid identity to the query sequence(s).1,3 The FLUture BLAST databases are populated with IAV sequences derived from submissions processed at the ISU-VDL, of which approximately one-half are confidential data. A user can submit multiple HA or NA sequences of mixed genes or multiple subtypes in either nucleotide or amino acid FASTA format (Fig. 1). Gene sequences can be processed by the multisequence identity tool as full or partial genomes and may contain ambiguities, although the accuracy of results may decrease if the sequences are degraded. Following submission, the H1, H3, N1 or N2 subtype, and phylogenetic clade of the IAV sequences with the highest nucleotide or amino acid identity determined using BLASTn or BLASTp (https://blast.ncbi.nlm.nih.gov/Blast.cgi), are presented graphically. In addition, metadata associated with the 10 ISU-VDL cases with ≥96% nucleotide or amino acid similarity to the query HA sequence are presented in tabulated and graphical form (Fig. 2A, 2B).
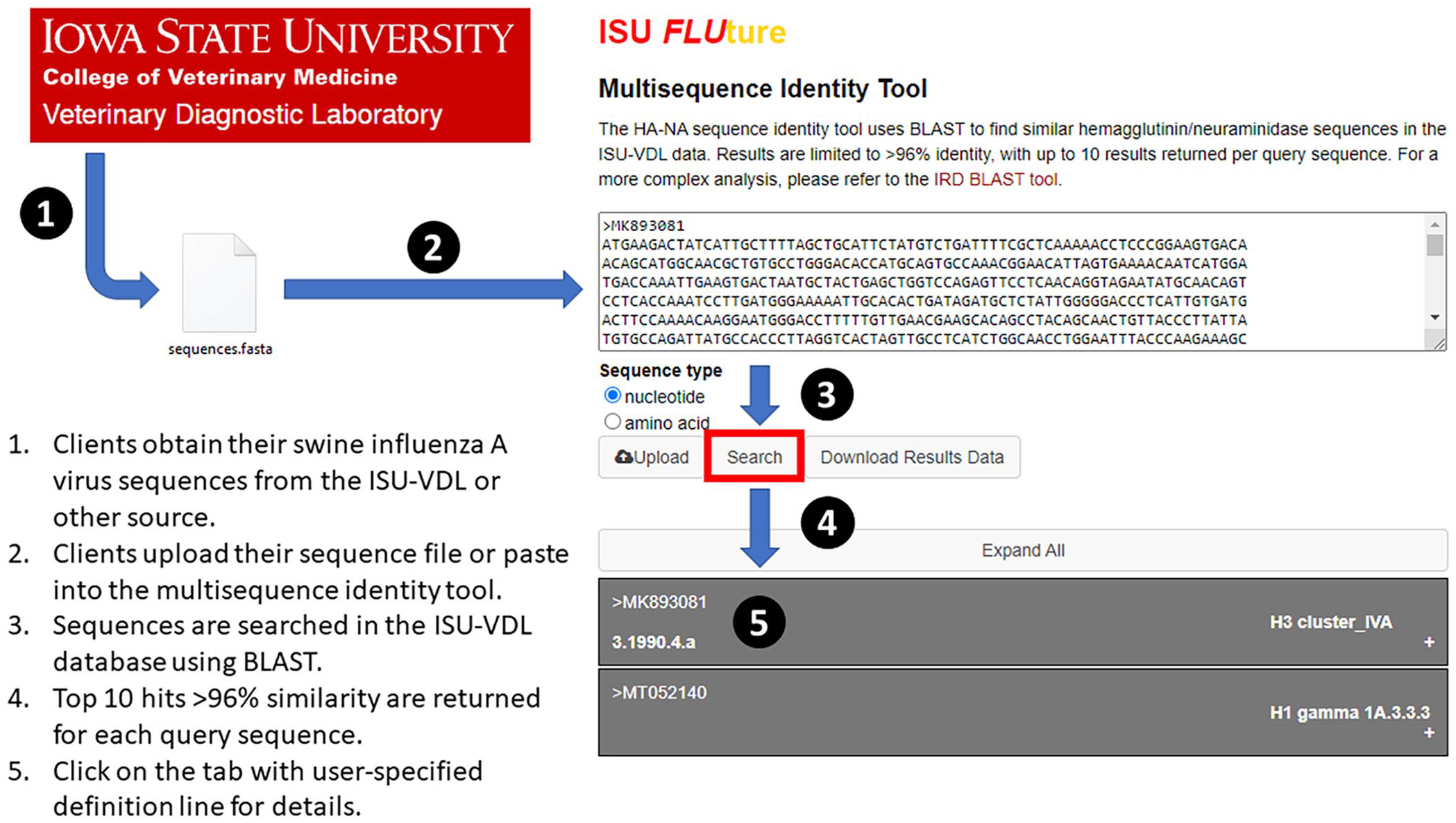
Flow chart of the process for using the ISU FLUture multisequence identity tool.
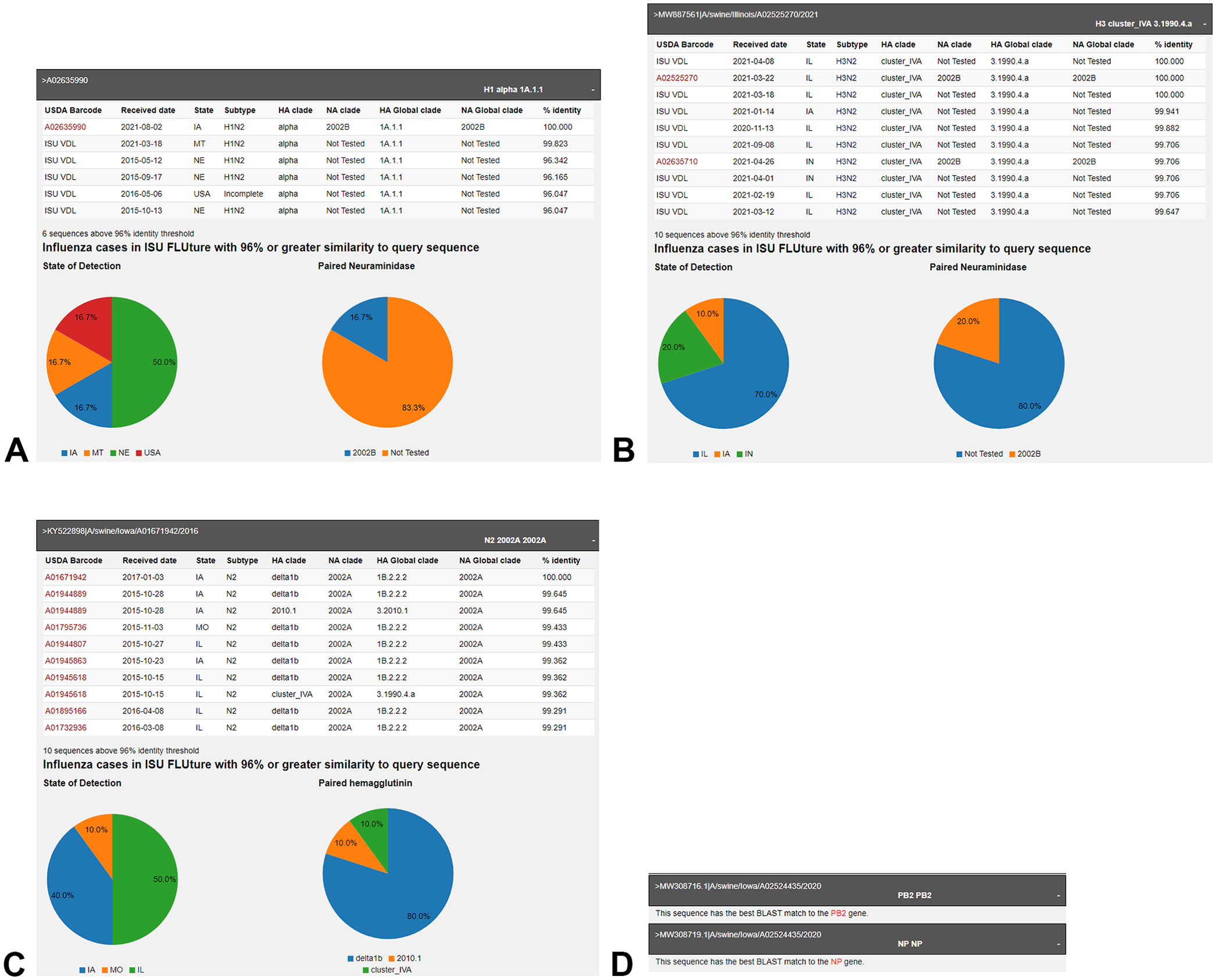
Query BLAST results provided by the ISU FLUture multisequence identity tool. Metadata associated with the top 10 sequences with ≥96% genetic identity within the Iowa State University Veterinary Diagnostic Laboratory (ISU-VDL) sequence repository populate a results table. Sequences included in the USDA surveillance program are identified with a barcode, the date the sample was submitted to the ISU-VDL, the U.S. state from which the case originated, and phylogenetic information.
Once the search is completed, a series of tabs labeled by the user-specified definition line of the FASTA query HA or NA sequence is returned. Each tab is annotated with the subtype as well as the phylogenetic clade based on U.S. and global swine clade designations 2 of the corresponding sequence with the highest percent identity. Each tab can be selected to expand a window that contains the additional metadata from the 10 sequences returned from the search. If identified sequences are in the public domain as part of the USDA IAV in swine surveillance system, a link is provided to the sequence in GenBank.2,3 USDA IAV surveillance sequences typically include HA and NA gene segments. The result for a query HA is a listing of the top 10 similar HA genes, and, if a paired NA sequence is available with the corresponding HA sequence, the NA phylogenetic clade is also provided. The U.S. state from which samples were collected is provided, or if not available, this is aggregated to a generic U.S. designation. Tabular results of the genetic similarity analysis can be downloaded into a CSV text file for reference and further use.
The critical functionality of our multisequence identity tool lies in determining the percentage of sequences with ≥96% similarity in the extensive ISU-VDL database. In addition to the tabular results, the graphical output of the query also includes percentage of detection by state summarized in a pie chart (Fig. 2A–C). These features allow quick assessment of the spatial distribution of genetically similar IAV sequences by state. A second pie chart visualizes the percent of which NA clades available in the ISU-VDL database are detected with similar HAs (Fig. 2A, 2B). Swine-origin NA sequences submitted as a query to the ISU FLUture multisequence identity tool provide output similar to the HA query, with the percentage of HA clades paired with the query NA presented in the pie chart (Fig. 2C).
The ISU FLUture multisequence identity tool databases are rebuilt weekly by adding newly classified sequences generated from diagnostic cases at the ISU-VDL. 17 Integration with our extensive diagnostic laboratory database provides the submitter a unique tool to identify whether the submitted sequence is similar to other genes. The ISU FLUture database has an additional 7,000 sequences with anonymized diagnostic data that are not published publicly because of client agreements. Additionally, curated reference sequences for non-HA/NA segments are maintained in the BLAST database and used to identify the presence of the 6 internal gene segments 1–3, 5, 7, and 8. 4 Because the database is comprised solely of ISU-VDL–derived data and because current IAV whole-genome sequencing efforts are limited at the ISU-VDL and at other veterinary diagnostic laboratories, more extensive analyses of IAV internal gene segments would need to be performed using other resources with more diverse databases, such as the Influenza Research Database (IRD, https://www.fludb.org).8,12 Results of BLAST searches are best interpreted if the user is aware of the submitted sequence origin and the limitations of genetic identity for biological inference. As an example, there are relatively frequent spillovers of human IAV into swine, particularly the H3 subtype.9,10,18 Given that some of these lineages, such as 2010.2, are relatively new to the swine population, they have not had time to diverge past the 96% identity criterion. A subsequent H3 human spillover event may then be incorrectly identified as an H3-2010.2 rather than being recognized as a novel spillover.
The BLAST databases are updated exclusively with swine IAV sequences that include case metadata that are submitted to the ISU-VDL. Use of the ISU FLUture tool should be limited to swine IAV sequences that are endemic to North America, namely gene segments that are derived from H1, H3, N1, and N2 subtype swine IAV, although internal gene segments from swine IAV can be submitted to the tool for identification only. Submission of query sequences of IAV endemic to other host species, or from international locations, are unlikely to return similar sequences from BLAST. In these cases, a more comprehensive BLAST search conducted in GenBank or through the IRD is more appropriate.8,12 Finally, for computational and visualization reasons, we restricted the presentation of tabular results to the top 10 hits of ≥96%: a consequence of this limitation is that the top 10 results may not be the best results or the most evolutionarily similar strains.7,11 A query that has hundreds of similar BLAST results will have the same number of results in the output as a query that only has 10 BLAST results.
A use-case for the tool occurs when a veterinarian submits clinical samples from a pig exhibiting influenza-like illness. Upon PCR detection of IAV, the submitter requests sequencing through private funding or submission to the USDA IAV in swine surveillance system. 3 Upon receiving the result, and sequenced HA gene(s), the submitter compares their data to determine whether the IAV genes representing the strains in their herd are unique or reflects a novel introduction into their region or production flow. Identification of a new HA or NA clade in a production system, detection of genetically divergent strains, or finding a clade not previously observed in a U.S. state warrants consideration of a vaccine update. Additionally, identification of different genetic clades of IAV strains in one production phase requires assessment of pig movement to minimize the risk of transmission and reassortment to the downstream production phase.
In our provided example, we demonstrate the detection of an H1 with only 6 hits ≥96% identity (Fig. 2A) and an H3 with ≥10 hits ≥96% identity (Fig. 2B). The search returned similar results from 5 states that had the most cases detected in 2021. If both viruses were derived from the same farm, a vaccine that minimally includes a similar H3 component could be recommended, given that this HA gene appears to be widely distributed and epidemiologically relevant. The H1 return requires additional study because this could represent the introduction of a novel H1 to the production system or migration of a strain from an underrepresented region. Clients who request NA sequencing or whole-genome sequencing may also submit these gene sequences to the tool. If a NA derived from the same farm was found to be N2 of the 2002A lineage (Fig. 2C), it may be advantageous to match the NA portion of a vaccine. If any of the 6 internal gene segments are submitted to the tool, the identification of the gene will also be returned (Fig. 2D).
Footnotes
Acknowledgements
We thank Siying Lyu for technical assistance.
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
Our work was supported in part by: the Iowa State University (ISU) Presidential Interdisciplinary Research Initiative; the ISU Veterinary Diagnostic Laboratory; the U.S. Department of Agriculture (USDA)–Agricultural Research Service (ARS project 5030-32000-120-00-D); the National Institute of Allergy and Infectious Diseases, National Institutes of Health, Department of Health and Human Services (contract 75N93021C00015); the Department of Defense, Defense Advanced Research Projects Agency, Preventing Emerging Pathogenic Threats program (HR00112020034); and the SCINet project of the USDA-ARS (project 0500-00093-001-00-D).