
Abstract
Rotaviruses (RVs) have been identified as one of the main infectious causes of diarrhea in young pigs. We determined the prevalence of rotavirus A (RVA), C (RVC), and H (RVH) in pigs on a Brazilian farm. Samples were screened by reverse-transcription (RT)-PCR, and samples positive for RVA were genotyped by PCR amplification and sequencing analysis. Of the 329 fecal samples analyzed, 102 (30.9%) were positive for RV, 25 (7.6%) contained RVA only, 32 (9.7%) contained RVC only, and 31 (9.4%) contained RVH only. Co-circulation, the presence of ≥ 2 RVs in a sample, was detected in 14 (4.2%) samples. Of the 15 animals with diarrhea, 6 (40%) were positive for RV, and of the 314 asymptomatic animals, 96 (30.6%) were positive for RV; there was no statistically significant difference between the 2 groups (p = 0.441). Genotyping of RVA strains showed co-circulation of genotypes G1, G3, G9-P[8]-I1, and I2-E1. Phylogenetic analysis showed that some of the RVA genotypes found in pigs had high percentages of identity when compared with reference strains from humans, which suggests interspecies transmission. Because RVs may be zoonotic, excretion of RVs into the environment can result in transmission to agricultural workers causing interspecies infections and allowing the emergence of new reassorted viruses.
Diarrhea is common, can result in dehydration, and is one of the leading causes of death in pigs. Diarrheal disease in pigs has a significant economic impact because it has a high mortality rate, changes feed conversion rates, and impacts weight gain leading to longer stays at the farm, which may further increase health problems. 19 Rotaviruses (RVs; Reoviridae, Rotavirus) are etiologic agents of acute diarrhea in several host species, including mammals and birds, and are classified into 12 species (A–L). 7 Rotavirus A (RVA) infections have been described in diarrheic and non-diarrheic animals worldwide and have had significant economic impacts on pig production. Rotavirus C (RVC) has been associated with diarrhea outbreaks and asymptomatic infections in pigs worldwide, and rotavirus H (RVH), an emerging pathogen, has been detected in pigs with and without diarrhea. 18 We investigated the prevalence of RVA, RVC, and RVH infections in pigs with and without diarrhea on a commercial farm in the state of Rio de Janeiro, Brazil, and characterized the RVA genotypes.
The fecal samples used in our study were harvested in November 2016 from a commercial farrow-to-finish pig farm in the municipality of Barra do Piraí, state of Rio de Janeiro, whose herd consists of ~ 3,000 animals. Fecal samples were collected from 329 pigs, aged 4–150 d, in the pre-weaning (≤ 20 d; n = 42) and post-weaning (21–150 d; n = 287) phases; 15 of the pigs had diarrhea (12 in the pre-weaning phase and 3 in the post-weaning phase), and 314 of the pigs did not have diarrhea (30 in the pre-weaning phase and 284 in the post-weaning phase). Our study was approved by the Animal Research Ethics Committee of the Federal University of Rio de Janeiro, Rio de Janeiro, Brazil (protocol 21/011).
Stool suspensions were prepared in 10% (w/v) phosphate-buffered saline (pH 7.2) and then centrifuged at 2,500 × g for 5 min; nucleic acid was extracted from 300 μL of the supernatant using the phenol–chloroform–proteinase K method. Specimens were tested for the presence of RVA, RVC, and RVH by reverse-transcription PCR (RT-PCR) amplification. For RVA testing, the primers VP6-Fdeg and VP6 were used for the first round of PCR, and the VP6-F1deg and VP6-R primers were used for semi-nested PCR. 13 RVC was detected using the protocol described previously. 5 For RVH testing, the primers 5F (5′-ATGGTTGCAGGGATTGAATCT-3′) and 5R (5′-TCCTTCAGTCATAACNGTTGATAC-3′) were used for the first round of PCR with a predicted product of 544 bp. Then, semi-nested PCR was performed with the primers 2F (5′-GCTGATGACCCATTTGCAAATTT-3′) and 5R to generate a 309-bp fragment of the viral capsid protein 6 (VP6) encoding gene. The PCR products were separated by 1.2% (w/v) agarose gel electrophoresis, stained with ethidium bromide, and visualized under UV light. A 100-bp DNA ladder (Ludwig Biotec) was used to determine molecular size.
Nine randomly selected PCR amplification products representing each of the surveyed RV species were sent to Macrogen (Seoul, Korea) for sequencing to confirm the results and to rule out possible contamination with positive control samples and/or primer cross-reactivity. The test nucleotide sequences were aligned with reference sequences available in GenBank to confirm the RT-PCR results.
RVA-positive samples were further analyzed by multiplex RT-PCR to determine the genotypes G (VP7), P (VP4), I (VP6), and E (NSP4, nonstructural protein 4). 13 To further validate the genotypes, samples were re-analyzed by singleplex PCR. To avoid cross-contamination, no positive controls were used for the PCR reactions; only negative controls were used to ensure the accuracy of the results.
To confirm the genotyping results, 39 select singleplex PCR amplification products were sequenced. Overlapping sequences were assembled and edited using SeqMan, EditSeq, and MegAlign in the Lasergene software package (DNASTAR). Finally, the sequences were compared with reference strain sequences from our laboratory collection. No cross-contamination was observed in our experiments. Phylogenetic analysis was performed with MEGA software (v.7.0.14; https://www.megasoftware.net/). Dendrograms were constructed using the maximum likelihood method based on the Kimura 2-parameter model. Statistical significance was estimated by bootstrap analysis with 1,000 pseudoreplicates. The sequences were compared to reference RVA strains from GenBank (https://www.ncbi.nlm.nih.gov/nucleotide/). Sequences generated in our study were deposited into GenBank under accessions MN201921–MN201957. Nucleotide identities were determined by using the MegAlign p-distance algorithm.
Of the 329 fecal samples analyzed in our study, 102 (30.9%) were positive for RV (Table 1), 25 (7.6%) contained only RVA, 32 (9.7%) contained only RVC, and 31 (9.4%) contained only RVH. Co-circulation, the presence of ≥ 2 RVs in a sample, was detected in 14 (4.2%) of the samples. Overall, RVA was detected in 9.1% of the samples, with 7.6% of the samples containing only RVA and 1.5% of the samples containing RVA and/or RVC and/or RVH. RVC was detected in 13% of the samples, with 9.7% of the samples containing only RVC and 3.3% of the samples containing RVC and/or RVA and/or RVH. RVH was detected in 13.4% of the samples, with 9.4% of the samples containing only RVH and 4% of the samples containing RVH and/or RVA and/or RVC. Of the 15 animals with diarrhea, 6 (40%) were positive for RV, and among the 314 asymptomatic animals, 96 (30.6%) were positive for RV. There was no statistically significant difference (p = 0.441).
Detection of rotavirus A, C, and H in fecal swine samples.

Numbers in parentheses are percentages.
Fourteen RVA-positive samples were genotyped (Table 2, Fig. 1). The results of multiplex PCR suggested co-circulation of multiple RVA genotypes. To confirm this, singleplex PCR was performed using specific primers for each identified genotype, the PCR amplification products were sequenced, and the results confirmed co-circulation of multiple RVA strains in the samples. However, cross-reactivity between genotypes G9 and G4 and between genotypes E1 and E3 was observed. We concluded the occurrence of cross-reactions when the sequences of the amplicons obtained with both primers for G9 and G4 or E1 and E2 identified only one genotype (G9 and E1). Nucleotide analysis showed that some of the pig RV genotypes shared a high percentage of identity with reference strains from humans, suggesting interspecies transmission (Table 3).
Identification of the G, P, I, and E genotypes of rotavirus A strains detected in swine in our study.
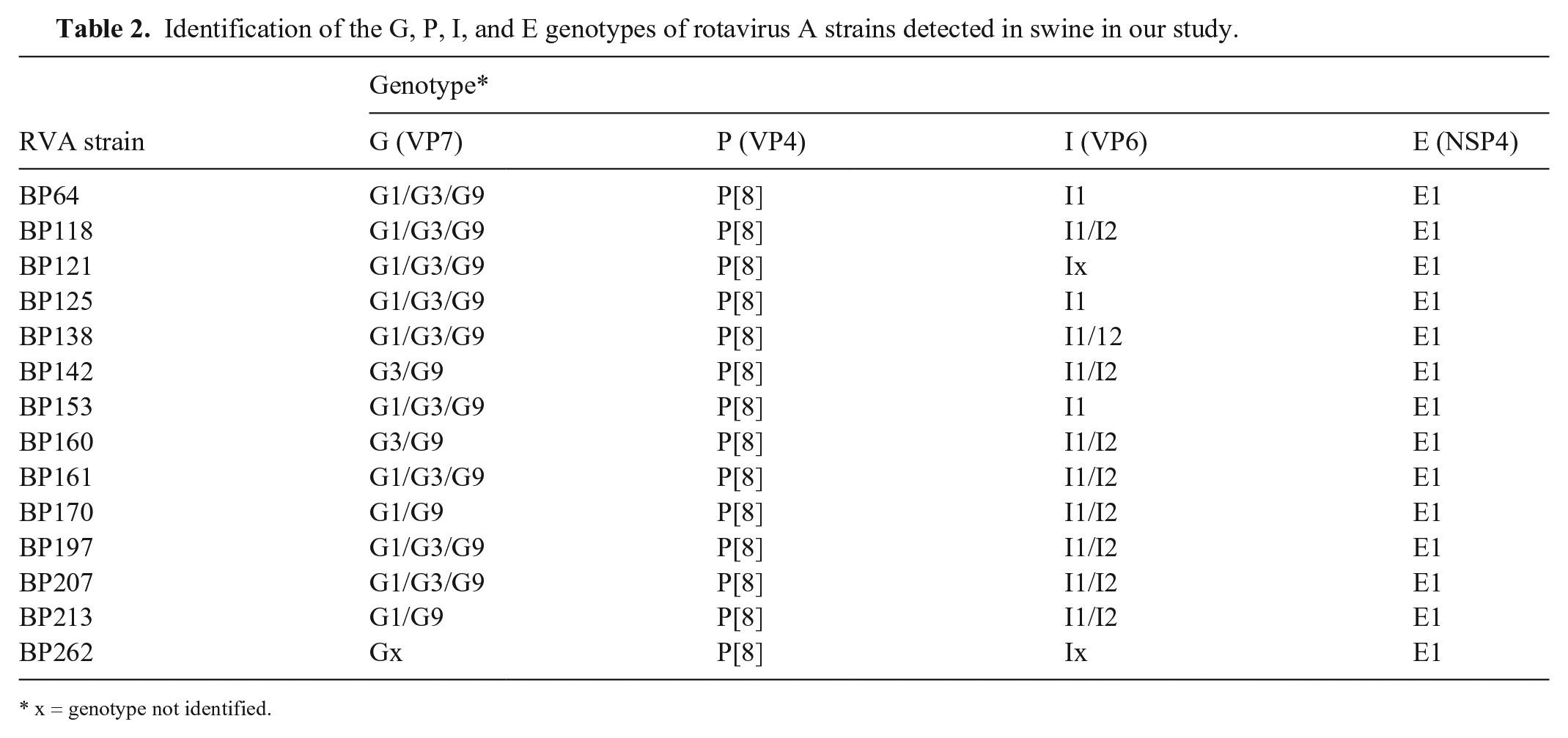
x = genotype not identified.
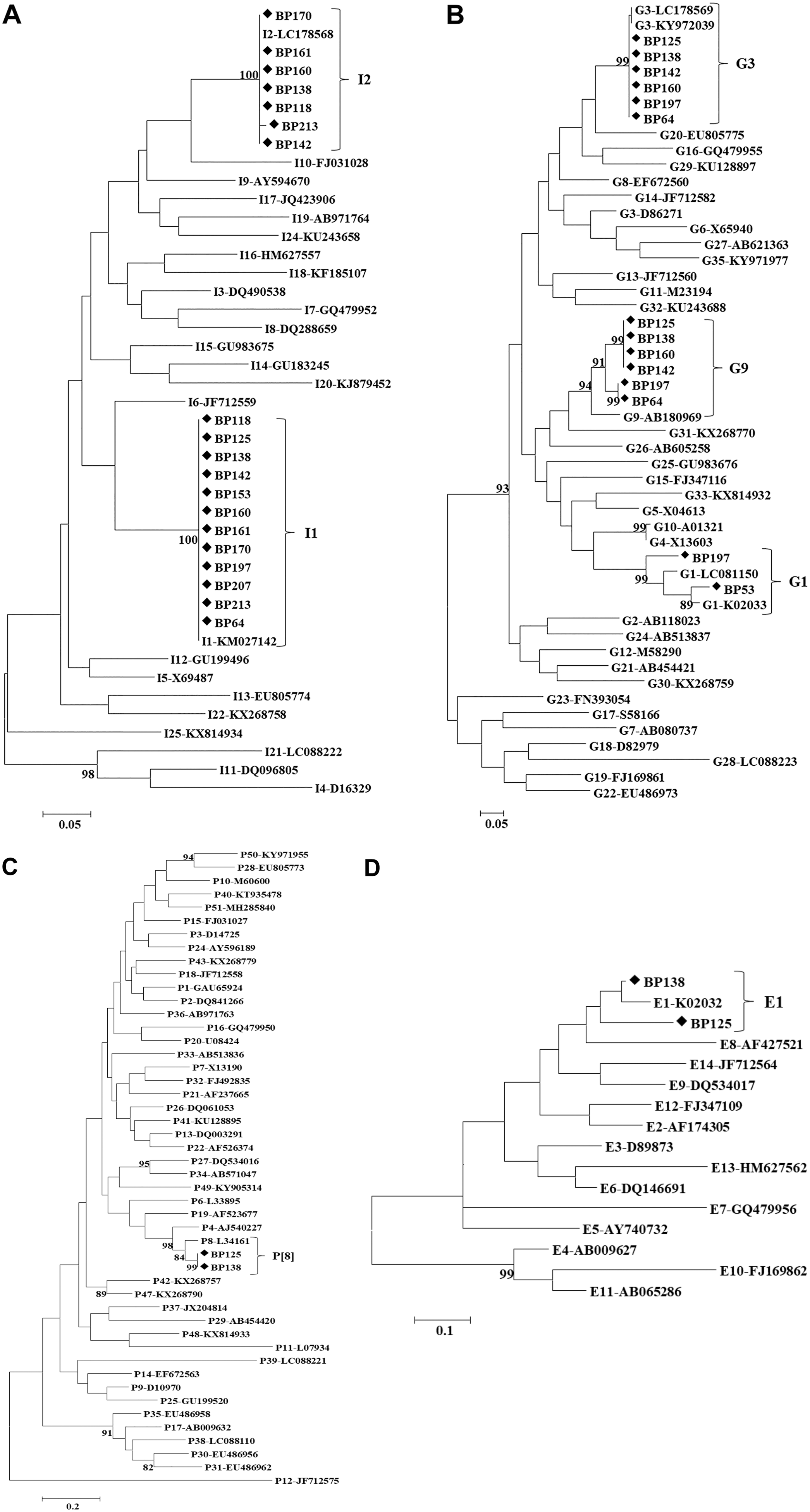
Phylogenetic trees constructed from partial nucleotide sequences of
Percentage of nucleotide identity of the genomic segments encoding VP6, VP7, VP4, and NSP4 of rotavirus A strains identified in our study compared to the nucleotide sequence of the cognate gene of the closest phylogenetically reference strains present in GenBank.
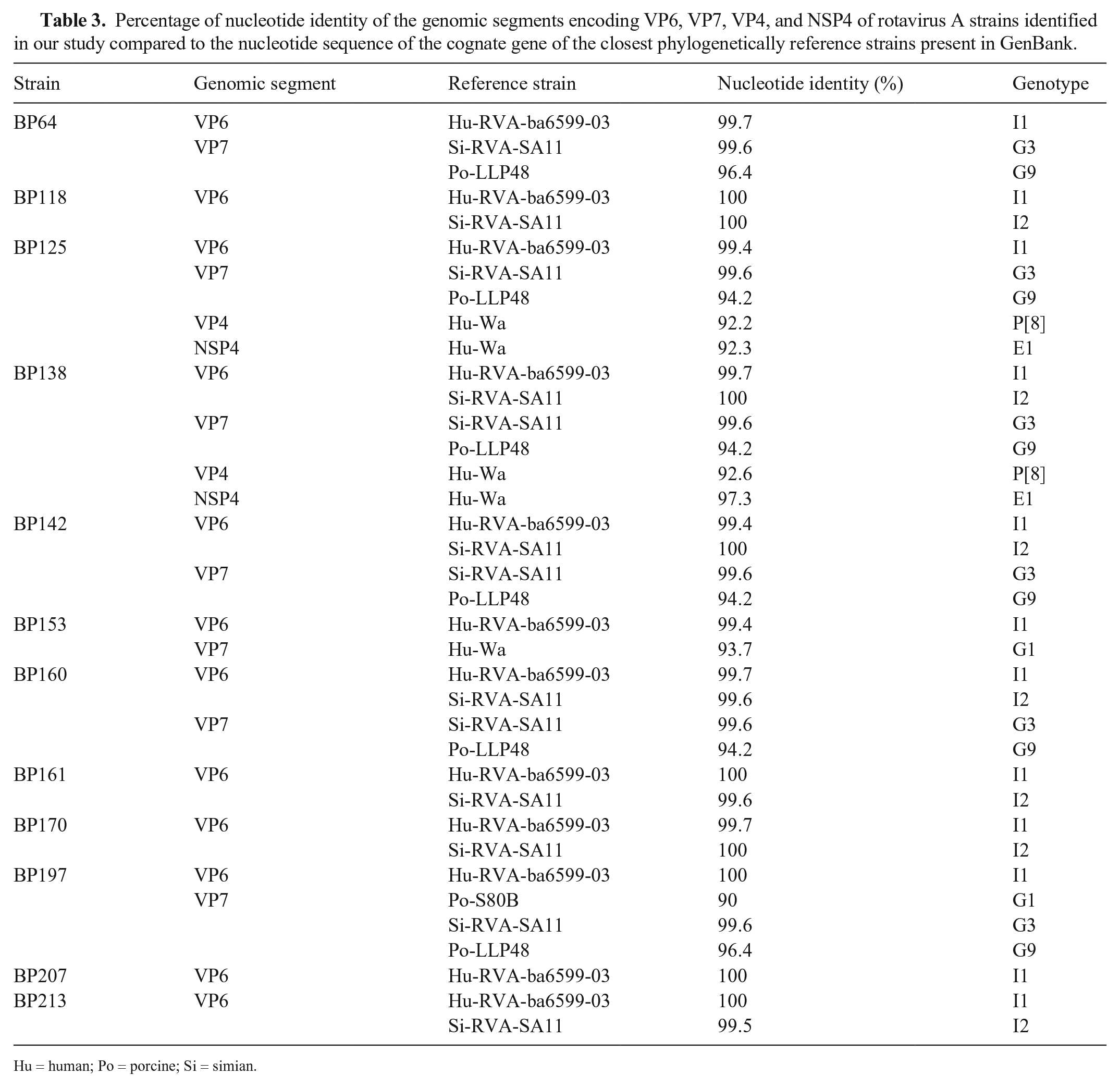
Hu = human; Po = porcine; Si = simian.
RVA infections with or without diarrhea have been described in pigs worldwide. 18 Epidemiologic studies have found a great diversity of RVA G and P genotypes in pigs and in humans,11,18 and a number of these genotypes are shared by humans and pigs, suggesting interspecies transmission.2,18 In general, RVA genotypes G3, G4, G5, P[6], P[7], and P[13] are found more frequently in pigs. Genotypes I1, I2, I5, I14, E1, and E9 have also been detected in pigs.8,11 Porcine genotypes G1–G5, G9–G11, G26P[6], P[7], P[8], P[13], P[23], I5, and E1 have been found in Brazil. 15
In our study, we identified RVA genotypes G1, G3, G9-P[8]-I1, and I2-E1. Porcine RVA genotypes G1, P[8], I1, and E1 had high nucleotide identities with cognate strains of human origin that are frequently detected in humans, 3 suggesting interspecies transmission. In agreement with this hypothesis, others 9 analyzed RVA strains in pigs in Spain and Italy and found G3, G4, G5, and G9-P[6]-I1-E1 genotypes with high nucleotide identities to human strains. In Taiwan, G9-P[19]-E1 strains were detected in both children and pigs. 20 Strain G11-P[6], which shares 90% identity with swine RVA, was detected in a child in Ecuador. 1 A study in Vietnam detected RVA strain G4-P[6]-I1-E1 in a pig and a human. 12 Monitoring of RVA strains circulating in Brazil revealed the presence of G5P[8] human strains with high degrees of identity to swine strains, and the presence of G1 and G5-P[8] swine strains with high degrees of identity with human strains.6,14 Less common genotypes may also circulate on farms, but they were not detected because primers specific for them were not used in the multiplex PCR reactions.
Interspecies transmission and gene rearrangements between homologous and heterologous strains are key factors in the evolution of RVA. 2 Thus, systematic surveillance of circulating strains in humans and animals is necessary to monitor the evolution of RVA and to identify the emergence of new and potentially pathogenic variants.
Interspecies RVC infections between swine and cattle have been described, but less frequently than interspecies transmission of RVA. The potential for transmission of RVC from pigs to humans was suggested after the detection of porcine-like RVC strains in children in Brazil. 4 In addition, porcine RVC strains containing genome segments of human origin have been identified. 8 Furthermore, complete genome analysis of RVC strains from swine in Japan showed genetic proximity to RVC strains from humans. 16 Full-genome analyses of RVC strains in pigs in the United States and Mexico suggested that pigs act as reservoirs for RVC and can be a source of infection for other species, including humans. 17 Thus, excretion of porcine RVC into the environment can lead to transmission to farmworkers, which allows for the emergence of new reassorted viruses.
RVH has been detected in humans, swine, and bats. 10 Sequence comparisons of the 11 genomic segments in various swine RVH strains showed that the nucleotide sequences were 81–94% identical and that the amino acid sequences were 81–100% identical. Sequence comparisons of pig and human RVH strains showed that the nucleotide sequences were 50–73% identical and that the amino acid sequences were 38–80% identical. Sequence comparisons of swine and bat RVH strains showed that the nucleotide sequences were 48–74% identical and that the amino acid sequences were 29–80% identical. 10 These data indicate that interspecies transmission of RVH has not been detected. 10
In our study, 30.6% of asymptomatic animals excreted RV. Four hypotheses could explain asymptomatic infections in these animals: (1) the persistence of maternal immunity; (2) because RV circulated widely on the study farm, animals could have been infected and developed immunity prior to sample collection, thus they would not currently have diarrhea; (3) clinical disease caused by RV is impacted by animal age and coinfections; and (4) because RV was detected by nested PCR, we do not know if the amount of RV shed by symptomatic and asymptomatic animals differs. Asymptomatic infection complicates efforts to identify and quarantine infected animals to prevent the spread of infection.
Our study reinforces the need to monitor asymptomatically infected animals because they contaminate the environment and can facilitate the spread of pathogens to the herd and to humans, especially farmworkers who have close relationships with the animals, which may allow the emergence of new reassorted viruses.
Footnotes
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
This study was supported by the Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq grants 404984/2018-5 and 301469/2018-0), the Fundação Carlos Chagas de Amparo à Pesquisa do Estado do Rio de Janeiro, Brazil (FAPERJ, grant E-26/202.909/2017), and Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES; Finance Code 001). The funders were not involved in the study design, data collection, data interpretation, or the decision to submit the work for publication.