
Abstract
We developed a SYBR green I–based reverse-transcription quantitative PCR (RT-qPCR) assay for bovine ephemeral fever virus (BEFV). Analytical sensitivity of the assay was ~ 100 times higher than conventional RT-PCR. The precision of the RT-qPCR established for RNA standards was high, with intra-assay and inter-assay coefficients of variation of 0.23–0.89% and 0.23–1.02%, respectively. The test was highly specific for BEFV strains, with no cross-reactivity with other viruses of veterinary significance. The assay detected BEFV RNA as early as 1 d post-infection (dpi) and up to 7–8 dpi in the blood samples of experimentally infected cattle. The most stable reference gene, peptidylprolyl isomerase A (PPIA), was selected for the quantification of BEFV. Viral RNA loads reached peak level at 3–5 dpi and then decreased rapidly through 7–8 dpi. Our assay provides a reliable approach for the detection of BEFV in the early infection stage and for use in the profiling of BEFV kinetics in vivo.
Introduction
Bovine ephemeral fever (BEF) is an economically important viral disease of cattle and water buffalo and is caused by bovine ephemeral fever virus (BEFV; Ephemerovirus, Bovine fever ephemerovirus). 24 The disease was first recorded in the 1910s, and the virus was isolated from cattle blood and several biting midges as well as mosquitoes in the 1960s and 1970s.23,24 BEFV infection in cattle usually results in high fever, reduced milk production, abortion, lameness or paralysis, and in some cases death. To date, BEF has been reported in more than 40 countries in Oceania, Africa, and Asia, causing considerable economic losses. 24
In addition to classical approaches, such as virus isolation and serologic assays, several molecular techniques have also been developed to detect BEFV infections including: RT-PCR, nested PCR in combination with magnetic bead–based DNA probing technology, reverse-transcription–loop-mediated isothermal amplification (RT-LAMP), and lateral-flow dipstick isothermal recombinase polymerase amplification (LFD-RPA).6,13,14,26 These methods have sufficient sensitivity to identify the virus but are either labor-intensive or do not allow direct quantification of viral RNA. As an alternative, TaqMan MGB–based quantitative RT-PCR (RT-qPCR) assays were also used for early detection of the BEFV genome.3,4,9,21
We developed a low-cost SYBR green I–based RT-qPCR assay using primers based on a conserved genomic region for BEFV strains originating from East Asia, the Middle East, and Australia. We evaluated the performance of the assay in an experimental BEFV infection study in Chinese Holstein cattle, in order to establish the earliest detection of the virus and the kinetics of the infection.
Materials and methods
Viral strains and plasmids
Chinese BEFV strain HN1/2012, previously isolated from dairy cattle in Henan province in 2012 and passaged 5 times by intravenous inoculation of calves, 11 was used for the experimental infection study. Additional BEFV strains LS11 and JT02L, 27 and the inactivated BEFV vaccine based on strain JB76H (Harbin Weike Biotechnology Development, Harbin, China), were used to evaluate the broad reactivity of the RT-qPCR. We tested the RT-qPCR specificity with several other viruses, including bluetongue virus 1 (BTV-1) strain GS/11, bovine viral diarrhea virus (BVDV) strain camel-6, recombinant vesicular stomatitis virus that carries a green fluorescent protein (GFP) reporter gene (VSV-GFP), live attenuated Japanese encephalitis virus (JEV) vaccine for swine (China Animal Husbandry Industry, Beijing, China), and rabies virus (RABV) inactivated vaccine (Intervet International, Boxmeer, The Netherlands). Recombinant plasmid pGEM-BEFVG containing the G gene of BEFV strain JB76H 27 was used for RNA standard development.
RNA extraction
Viruses and vaccines (100 μL) were used for RNA extraction (QIAamp viral RNA mini kit; Qiagen, Hilden, Germany) with on-column DNA digestion. The extracted RNA was eluted with 30 μL of nuclease-free water and stored at −80°C until use.
Primer design
Representative G gene sequences of BEFV strains from East Asia, the Middle East, and Australia were retrieved from GenBank and aligned with the MEGA 6.0 program. A pair of specific primers were designed from a relatively conserved region (nucleotide position: 1640-1840) with a product of 201 base pairs (bp) for the SYBR green I–based RT-qPCR. The forward primer 1640F (5′-TAGTCACMACVTATGCDATYTAC-3′) and the reverse primer 1840R (5’- GTTTACTYCCACYYTTGATGCT-3′) were synthesized by GenScript (Nanjing, China); primers 420F (5′-GAGCTTGGTGTGA ATAC-3′) and 420B (5′- CCAACCTACAACAGCAGATA-3′) for conventional RT-PCR 26 were acquired from the same company. To normalize and quantify BEFV RNA in the infected cattle, we used primers for the candidate reference genes including tyrosine 3-monooxygenase/tryptophan 5-monooxygenase activation protein zeta (YWHAZ), succinate dehydrogenase complex subunit A (SDHA), 12 peptidylprolyl isomerase A (PPIA), 5 C-terminal binding protein 1 (CTBP1), RAD50 double-strand break repair protein (RAD50), 8 glyceraldehyde-3-phosphate dehydrogenase (GAPDH), and actin beta (ACTB). 17
Preparation of RNA standards
A 1,786-bp fragment of the G gene of BEFV strain JT02L was amplified with primers 76F (5′- TGTGGAGAACTTCATCCTGTA-3′) and 1861R (5′-AGAATCTATCATCACCGATTGG-3’) and cloned into pGEM-T easy vector (Promega, Beijing, China). The recombinants were screened with the M13R (5′-CAGGAAACAGCTATGAC-3′) and 1861R primers, followed by sequencing to confirm the direction of the insert. The obtained plasmid pGEM-BEFGR was linearized by NotI and subsequently used for RNA transcription (TranscriptAid T7 high yield transcription kit; Thermo Fisher Scientific, Shanghai, China) to obtain the RNA standards homologous to the negative-sense single-stranded RNA genome. The synthesized RNA was digested (RNase-free DNase; TaKaRa, Dalian, China), followed by extraction with phenol-chloroform and precipitation with 70% ethanol. The RNA pellet was dissolved with RNase-free water. The RNA concentration was determined (NanoDrop 2000 spectrophotometer; NanoDrop Technologies, Montchanin, DE), and the copy number was determined using the formula: copies/μL = 6.02 × 1023 [concentration (ng/μL) × 10-9/molecular weight (g/mol)] (http://www.endmemo.com/bio/dnacopynum.php). Subsequently, the RNA was serially 10-fold diluted from 1011 copies/μL to 101 copies/μL using easy dilution buffer (TaKaRa) and used as RNA standards.
SYBR green I–based RT-qPCR
One-step RT-qPCR was set up (One Step SYBR PrimeScript PLUS RT-PCR kit; TaKaRa) in a 25-μL format with 12.5 μL of 2× One Step SYBR RT-PCR buffer, 0.5 μL (10 μM) of each forward and reverse primer, 0.5 μL of ROX reference dye II (50×), 8 μL of nuclease-free water, 1.5 μL of ExTaq HS mix, 0.5 μL of RTase mix, and 1 μL of RNA. The reaction was performed (Mx3005P qPCR system; Agilent Stratagene, Santa Clara, CA), with reverse transcription at 42°C for 20 min, denaturation at 95°C for 30 s, 40 cycles of 95°C for 10 s, and 60°C for 30 s. Melting curve and standard curve analysis were performed according to the instructions of the qPCR system. To validate the reproducibility of the assay, each standard dilution was tested in triplicate to calculate the intra-assay coefficients of variation (CVs) of Ct (cycle threshold) values. Additionally, the standard was run 3 times on different days to evaluate the inter-assay CVs of Ct values. The RNA extracted from individual BEFV strains, BTV-1, BVDV, VSV-GFP, JEV vaccine, and RABV vaccine were used for evaluating the broad reactivity within species and the inter-species specificity of the assay, respectively. For sensitivity comparison, each RNA standard dilution was tested by the conventional one-step RT-PCR as described previously. 26
Experimental infection
Six clinically healthy male Chinese Holstein cattle, 12–14 mo old, were included in the experiment. All procedures were approved by the Animal Ethics Committee of Lanzhou Veterinary Research Institute, CAAS (LVRIAEC2013-010). Prior to infection, the animals were confirmed to be negative for anti-BEFV antibodies by indirect ELISA 15 and viral nucleic acid by RT-PCR. Baseline body temperatures were recorded for 3 consecutive days prior to exposure. The young bulls were randomly divided between the infected and control groups, with 3 animals in each group, and were maintained in facilities free from biting insects. Based on previous data from animal experiments, 28 8 mL of blood from calves infected with strain HN1/2012 was inoculated intravenously into each animal in the infected group; the animals in the control group received no treatment. Rectal temperatures were measured twice daily until they returned to the baseline level. Stabilized whole blood samples and serum samples were collected by venipuncture on the pre-inoculation day and then by daily blood sampling for 10 d post-inoculation (dpi). Further, serum samples for antibody detection were collected at 14, 21, 30, 60, 90, 120, 150 dpi.
BEFV detection and seroconversion in the inoculated cattle
Serum samples were tested for specific BEFV antibodies. 28 Isolation of BEFV from the uncoagulated whole blood samples was attempted by sequential passages in Aedes albopictus (C6 /36) cells and BHK-21 cells, 20 followed by detection with our RT-qPCR and the indirect fluorescent antibody assay described previously. 25 RNA was extracted directly from 1 mL of each EDTA-stabilized whole blood sample (QIAamp viral RNA mini kit; Qiagen). Total RNA was eluted with 20 μL of elution buffer and detected with RT-qPCR. Moreover, for the SYBR green I–based RT-qPCR, 6 candidate reference genes (including YWHAZ, SDHA, PPIA, CTBP1, RAD50, GAPDH, and ACTB) were evaluated for the sample-to-sample variations in the clinical blood samples. The amplification of each reference gene mRNA was set up similarly as the assay for BEFV detection and performed in duplicate with RNA from blood of the 3 BEFV-negative cattle in the control group and the 3 BEFV-infected individuals. The candidate reference genes were evaluated for stability by BestKeeper v.1, 16 NormFinder v.0.953, 1 and geNorm v.3.5. 22 The most stable reference gene(s) was used to normalize the BEFV RNA levels in an individual sample, using the 2-DeltaCt method. 18
Results
Analytical specificity of the RT-qPCR
The specificity of the assay was confirmed for all of the BEFV reference strains used: JB76H, JT02L, LS11, and HN1/2012. A melting temperature (Tm) of 79.8 ± 0.1°C was specific for the G gene fragment amplified in all of the samples. No specific signal was detected in the samples containing BTV-1, BVDV, VSV-GFP, JEV, and RABV (Supplementary Fig. 1).
Analytical sensitivity of the RT-qPCR
A standard curve based on the Ct values of a series of in vitro transcribed BEFV RNA was constructed, demonstrating dynamic linearity with a high regression coefficient of determination (R2 = 0.999). The BEFV RNA standard was reliably amplified down to a lower limit of 10 copies per reaction (Fig. 1). By contrast, the conventional RT-PCR was 100× less sensitive, with the last detectable dilution containing 103 viral RNA copies/μL (Fig. 2).
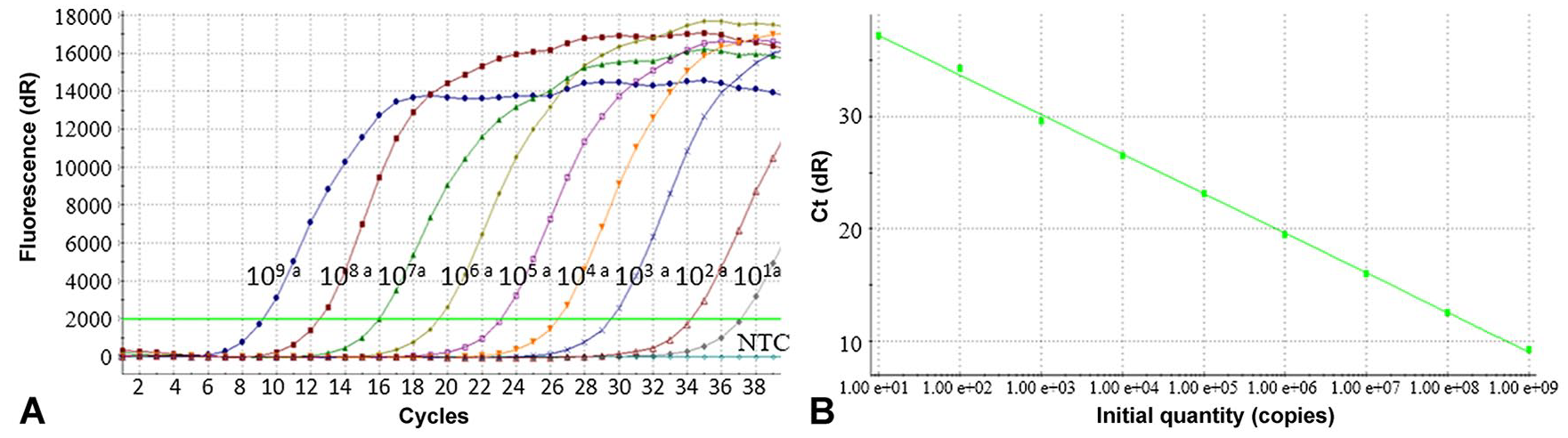
RT-qPCR detection of bovine ephemeral fever virus RNA standards.
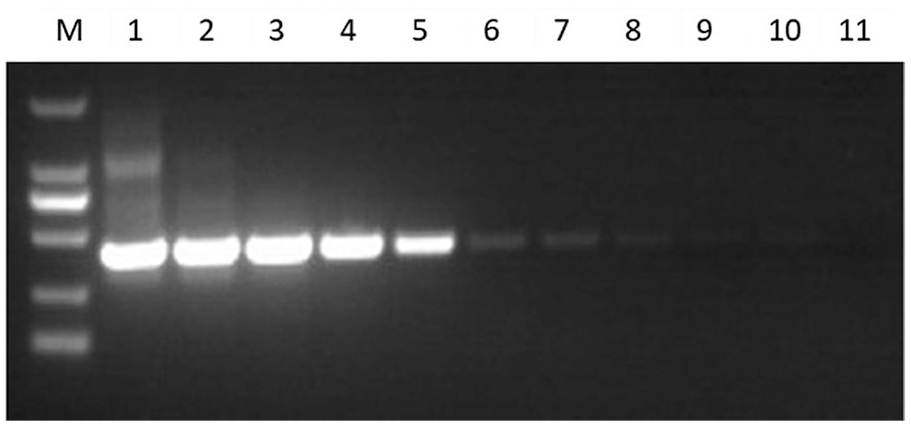
Detection of the bovine ephemeral fever virus RNA standards with conventional RT-PCR.
Reproducibility of the RT-qPCR
The Ct values of 6 RNA standards from 101 to 106 copies were compared to determine the intra- and inter-assay reproducibility. The CVs in intra-run and inter-runs were 0.234–1.02% (Table 1) and 0.208–1.02% (Table 2), respectively, indicating that the assay had very little variation.
Intra-assay reproducibility of Ct values in the RT-qPCR for bovine ephemeral fever virus.
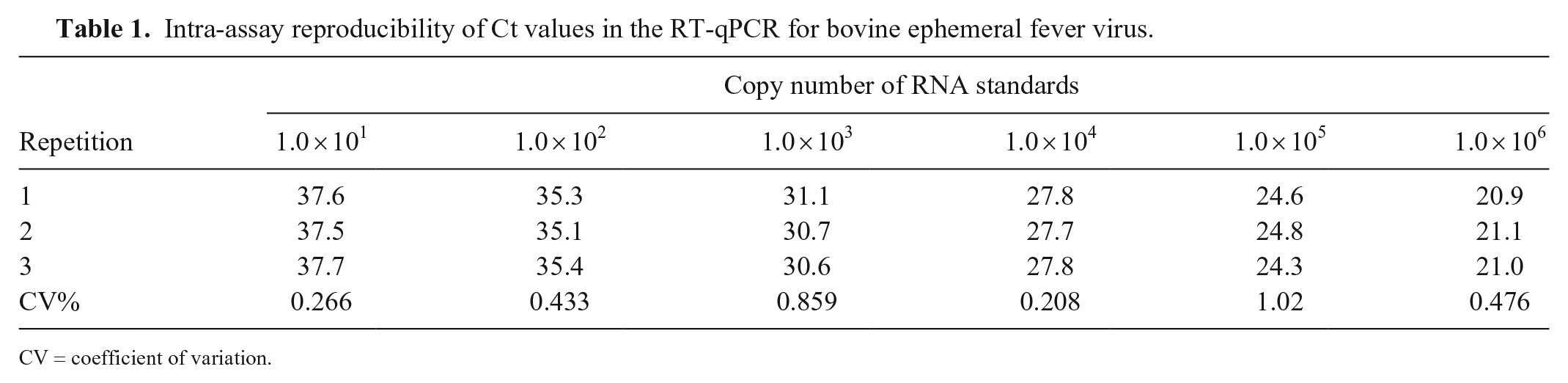
CV = coefficient of variation.
Inter-assay reproducibility of Ct values in the RT-qPCR for bovine ephemeral fever virus.
CV = coefficient of variation.
Clinical observations and serologic findings
The animals in the control group remained clinically healthy and had no increase of body temperature throughout the study period. All 3 of the animals in the infected group had increased rectal temperatures of 39.0–40.2°C at 3 dpi. A marked increase in rectal temperature was observed between 4 and 5 dpi in 2 animals from the infected group (046 and 001), with a maximum temperature of 41.7°C observed in calf 046 at 5 dpi. The increased rectal temperatures in the infected group were significantly higher than in the control group (p < 0.05). The fever was transient and lasted 3 d in animals 046 and 001, and 4 d in animal 002 (Fig. 3). Additionally, clinical signs of nasal and eye discharge were observed at 4–5 dpi in the infected group. Two animals (046 and 001) seroconverted as early as 4 dpi. The sample-to-positive control ratio (S/P) values peaked at 21–30 dpi, followed by a gradual decline until 120 and 150 dpi, when no antibodies were detected. In animal 002, BEFV antibodies appeared at 7 dpi and peaked 9 d later, but the S/P value was slightly lower than in the remaining 2 animals of the group. The antibody level declined gradually until 90 and 120 dpi (Fig. 4). The serum samples from the control group remained seronegative throughout the study.
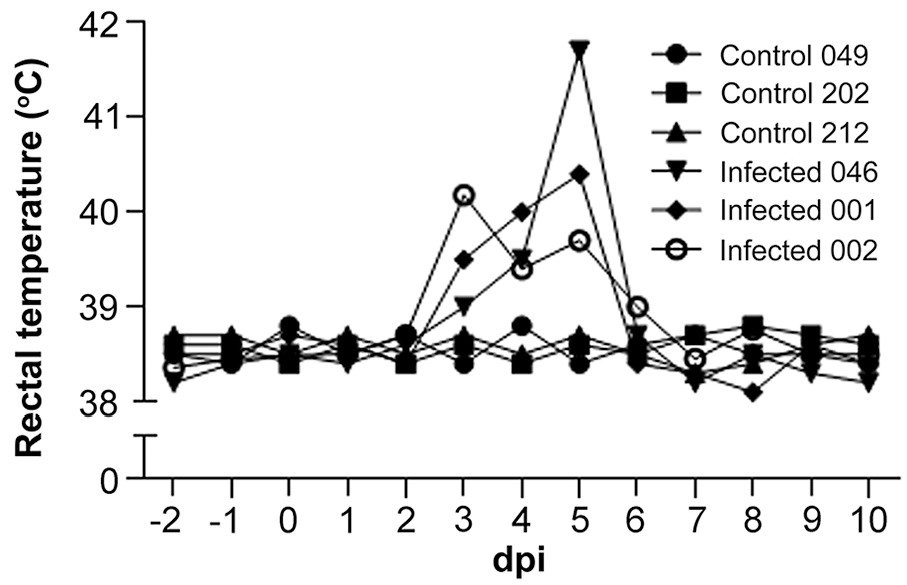
Rectal temperature (°C) in cattle. dpi = days post-infection.
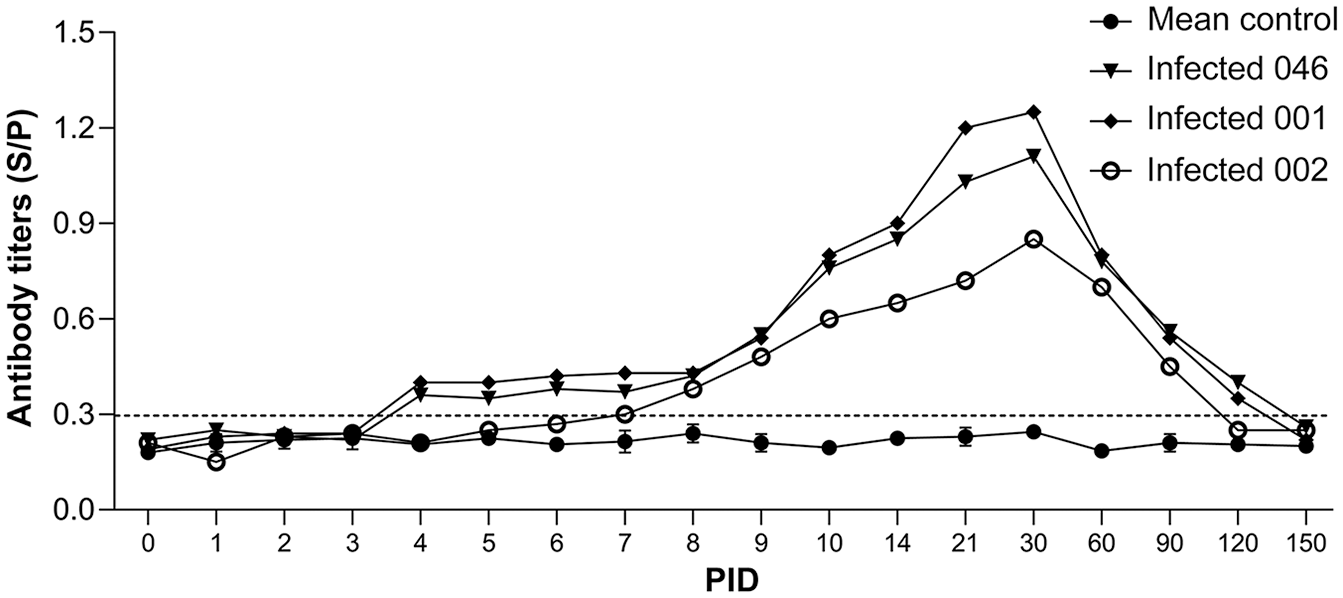
Dynamics of antibody measured by indirect ELISA.
Viral kinetics
All of the blood samples from the control group remained negative for BEFV by isolation and for viral RNA by PCR. BEFV was isolated in A. albopictus (C6 /36) cells from whole blood samples collected from animals 046 and 001 at 3–6 dpi, as well as from animal 002 at 3 and 4 dpi. BEFV genomic RNA was detected in the blood of infected animals at 1–8 dpi (Fig. 5A, 5B). Using blood samples from both groups, the candidate reference genes can be divided into categories including least abundant (YWHAZ, SDHA, RAD50), moderately abundant (CTBP, PPIA, GAPDH), and highly abundant (ACTB) according to the Ct values obtained. PPIA was consistently the most stable among reference genes in blood samples as assessed by geNorm and NormFinder. It also ranked first with the best coefficient of correlation (r; p = 0.001) by BestKeeper (Table 3). Therefore, PPIA was selected to normalize the BEFV RNA levels in individual samples. In animals 046 and 001, the level of viral RNA in the peripheral blood on the first 2 d post-inoculation increased rapidly at 3 dpi. Peak levels of viral RNA with a 104–105 increase were observed at 4 and 5 dpi, followed by a decrease through 7 and 8 dpi, until they dropped below the detection limit of RT-qPCR. By contrast, in animal 002, the peak of viral RNA was lower, but appeared earlier and displayed a slower decline after the peak (Fig. 5A). The kinetics of the RNA curves of raw Ct values did not differ significantly from normalized using 2-DeltaCt. Peak viral RNA levels in the blood were 3.08 × 105 copies/mL for bull 002 at 3 dpi and 1.45–2.01 × 107 copies/mL for bulls 046 and 001 at 5 dpi (Fig. 5B).
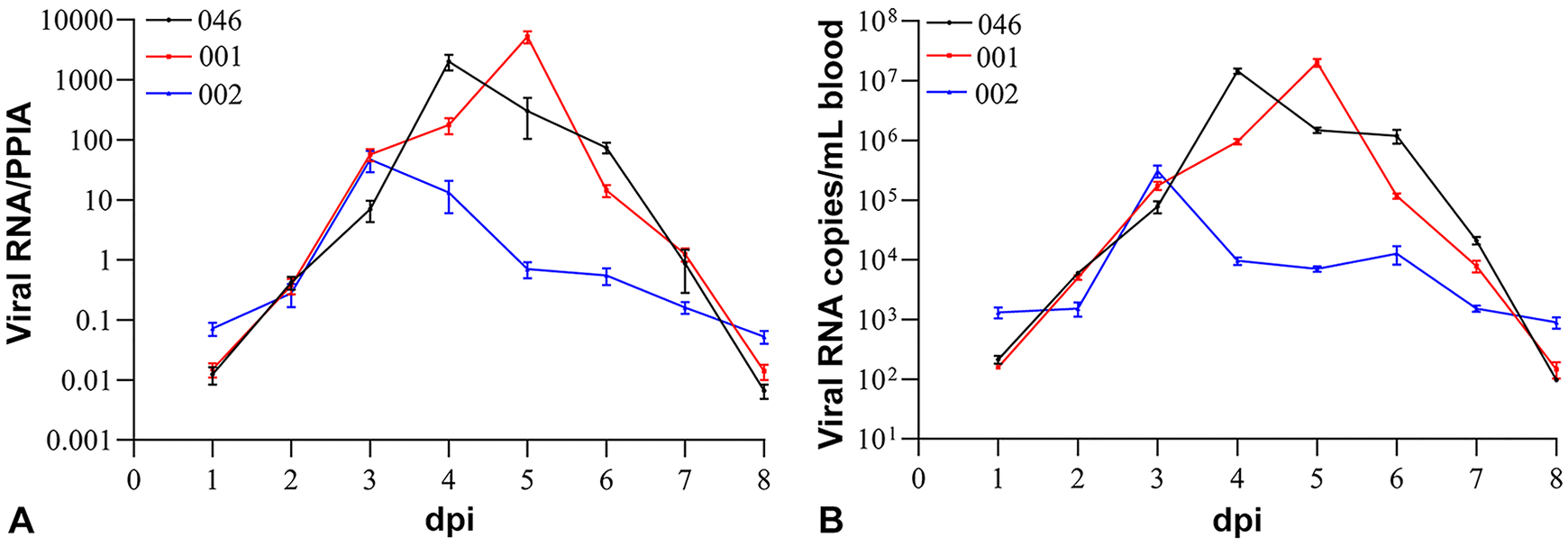
Kinetics of bovine ephemeral fever virus RNA in the blood.
Stability rankings of the reference genes for normalizing the bovine ephemeral fever virus RNA according to geNorm, NormFinder, and BestKeeper.
Discussion
Fluorescent probe–based RT-qPCR has been used for early detection of BEFV in cattle.4,5,9 However, probe-based RT-qPCR assays were developed based on Israeli and Australian BEFV isolates, and decreased sensitivity with a detection limit of ~100 copies was observed in our laboratory, which may be caused by mismatches of the probe sequence (data not shown). We therefore developed an inexpensive SYBR green I–based qRT-PCR assay for BEFV detection. Degenerate nucleotides were introduced into the primers to account for sequence variation of the G gene, making it possible to detect a wide spectrum of BEFV strains. Although the degenerate primers can result in a lower effective concentration of each individual primer, the analytical sensitivity of the assay was found to be comparable to a TaqMan probe–based qRT-PCR assay described previously. 21
Our RT-qPCR proved to be useful in quantitative analysis of viral RNA kinetics in Chinese Holstein cattle experimentally infected with BEFV stain HN1/2012, which belongs to a novel sublineage of the East Asia lineage. 11 The infection was confirmed by clinical observations, virus isolation, and the presence of specific antibodies. To avoid quantifying errors arising from variations in the quantity of the RNA samples, we investigated the suitability of reference genes for normalizing the viral RNA levels. In agreement with previous studies,7,8,19 we found that the commonly used ACTB and GAPDH genes were not stably expressed in BEFV-infected bovine whole blood samples, although they were respectively identified as stable reference genes in BVDV-infected bovine lymphoid cells 2 or Madin–Darby bovine kidney cells. 10 CTBP1 and RAD50 have been found stably expressed in bovine blood mononuclear cells 8 or bovine peripheral lymphocytes, 19 but neither was stable in our study. SDHA was previously reported as one of the most stable genes in bovine preimplantation embryos. 12 Similarly, it was ranked in our study as the second most stable reference gene by geNorm and NormFinder. In addition, YWHAZ and PPIA were previously found stable in bovine peripheral lymphocytes 19 ; we obtained a similar finding for PPIA in our study, except that YWHAZ was identified as the least stable gene during BEFV infection. Although discrepancy in the ranking orders of reference genes was observed by different software, PPIA was consistently identified as the most stable reference gene. The relative quantification of BEF viral RNA on each dpi resulted in more accurate and reliable profiling of viral kinetics in cattle.
Supplemental Material
Supplemental_material – Supplemental material for A SYBR green I–based quantitative RT-PCR assay for bovine ephemeral fever virus and its utility for evaluating viral kinetics in cattle
Supplemental material, Supplemental_material for A SYBR green I–based quantitative RT-PCR assay for bovine ephemeral fever virus and its utility for evaluating viral kinetics in cattle by Shandian Gao, Junzheng Du, Zhancheng Tian, Qingli Niu, Dexuan Huang, Jidong Wang, Jianxun Luo, Guangyuan Liu and Hong Yin in Journal of Veterinary Diagnostic Investigation
Footnotes
Acknowledgements
We thank Dr. Jingfeng Wang from Suzhou Institute of Systems Medicine, Chinese Academy of Medical Sciences for providing the recombinant vesicular stomatitis virus (VSV-GFP). We thank Dr. Fuying Zheng from Lanzhou Veterinary Research Institute, Chinese Academy of Agricultural Sciences for supplying the BEFV strains and for assistance in isolating the BEFV. We also thank Dr. Magdalena Larska (National Veterinary Research Institute, Poland) for her kind help in revising the manuscript.
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
This study was financially supported by the Special Funds for Agro-Scientific Research in the Public Interest (201303035); Central Public-interest Scientific Institution Basal Research Fund; ASTIP, CAAS, and NBCITS, MOA (CARS-37).
Supplementary material
Supplementary material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.