
Abstract
Bovine respiratory syncytial virus (BRSV) is circulating in cattle in Europe. Although vaccination helps control the disease, its prevalence within and among herds remains high. Previous genetic characterization studies revealed a strict geographic correlation between viral variants; on the other hand, they showed the emergence of new variants in northern Europe. Few studies have described BRSV distribution, and little is known about the genetic features of BRSV strains circulating in Italy. We studied sample-positive tests for BRSV, and sequenced the coding regions of the G and N proteins to determine the presence of divergent variants. Two different sets of sequences were found, including in samples from animals from vaccinated herds. The 2 groups of sequences correspond to 2 time periods and suggest an active role of herd immunity in preventing the spread of infection. Our findings that different strains of BRSV are circulating in Italy and that the virus is evolving rapidly highlight the importance of updating vaccination strategies.
Since the end of the 1960s, bovine respiratory syncytial virus (BRSV; species Bovine orthopneumovirus, genus Orthopneumovirus, family Pneumoviridae) has caused an acute respiratory disease syndrome in beef and dairy calves 13 and regular winter outbreaks of respiratory disease in cattle. 18 BRSV is distributed worldwide, and its impact on the cattle industry is associated with economic losses as a result of morbidity, mortality, costs of treatment and prevention, loss of production, and reduced carcass value. 16 Although BRSV is transmitted primarily by direct contact with infected animals or by aerosol, 11 its transmission can also be influenced by biotic and abiotic risk factors. 12
The presence of maternally derived antibodies is known to pose a major obstacle to efficacious vaccination. This problem may now be overcome, 1 but vaccine failure could be at least partially attributed to a possible broader antigenic spectrum of the BRSV population. Like most RNA viruses, BRSV has high genetic heterogeneity and a rapid evolutionary rate 15 forming different viral subpopulations within a single host. The complex mixture of viral variants, called quasispecies, can lead to divergent strains. This viral feature is particularly important in relation to the efficacy of BRSV prophylaxis.
The G viral protein has been identified as the major attachment protein, given that antibodies specific to the G protein were shown to block binding of the virus to cells. 10 Owing to its genetic and antigenic heterogeneity, the G protein, together with the nucleoprotein (N protein) and the fusion (F) protein, has been used as a target to better classify the viral strains of BRSV. 17
Several studies have revealed the high prevalence of BRSV both within and among herds in Europe.7,6,20 Moreover, genetic characterization studies have reported a strict geographic correlation between viral variants and the emergence of new variants in northern European countries 17 since the late 1990s.
The few studies published on BRSV distribution in Italy have focused on wildlife,3,5 and little is known about the genetic features of BRSV strains circulating in cattle herds. We studied samples positive for BRSV to identify circulating viral strains and to determine the presence of new variants. We selected a sample set from among the samples tested by the Istituto Zooprofilattico Sperimentale dell’Umbria e Marche (IZSUm) diagnostic laboratory, including specimens from BRSV outbreaks throughout Italy that had occurred in 2012–2015 (Table 1). Positivity to BRSV was determined using a real-time PCR assay described previously, 19 and by targeting the gene encoding glycoprotein F.
Samples used for study of bovine respiratory syncytial virus in Italy.
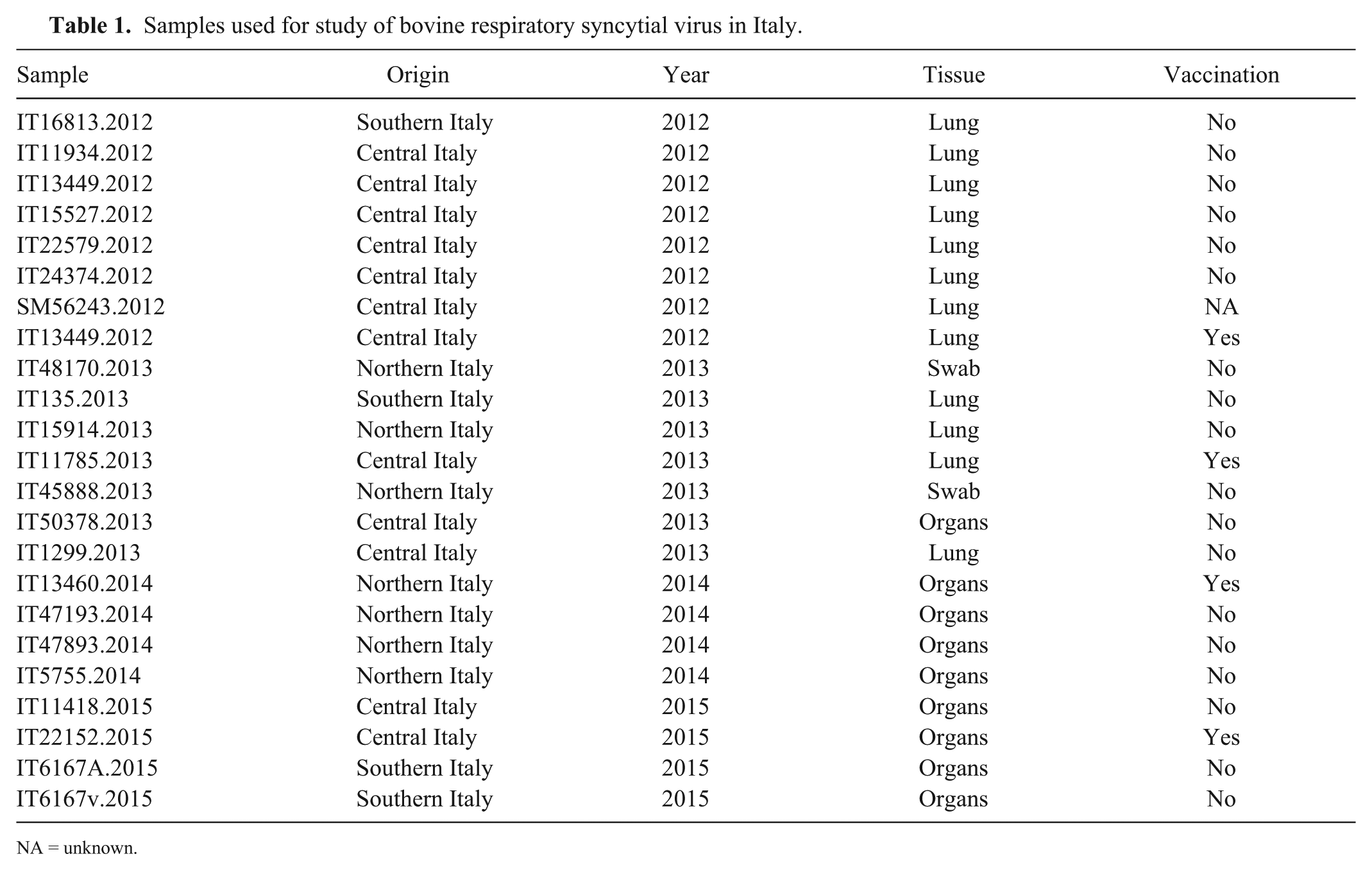
NA = unknown.
RNA was extracted (Qiagen EZ1 virus mini kit, Qiagen, Hilden, Germany), and eluted RNA was used as a template for amplification of the G coding sequence. Amplification was performed (Qiagen One-step RT-PCR kit, Qiagen) applying the nested protocol previously published 17 (Supplementary Table 1), following the manufacturer’s instructions.
After the first amplification step (primer pairs G2.5-F2.7 and N2.1-N2.2; Supplementary Table 1), PCR results were checked by agarose electrophoresis; samples showing the expected band (~1kb) were directly sequenced. The nested protocol (primer pairs VG1-VG4 and N2.3-N2.4) was applied only to the samples that did not test positive after the first amplification cycle. A set of G sequence–positive samples was used in amplifying a partial region of the N protein to confirm the subgroup association.
All PCR-positive samples were sequenced in both directions (BMR Genomics, Padua, Italy), and the electropherograms were checked manually. A set of reference sequences was selected from GenBank, including the 6 subgroups proposed previously. 17 Sequences were aligned with respect to the coding frame, and genetic heterogeneity was evaluated. The uncorrected p distance was calculated for the samples, and a phylogenetic tree was drawn by applying the best evolutionary model selected by the jModelTest 4 and the Bayesian approaches included in the MrBayes v.3.2.5 software. 14 Evolutionary rate was evaluated using BEAST v.2.4.3 software. 2
Sequence analysis revealed the presence of different BRSV strains circulating in Italy. For the G protein gene sequence (Fig. 1), 2 samples (IT111418-2015 and IT48170-2013) were from non-vaccinated farms and were strictly related to old subgroup III, similar to vaccine strains such as the FS-1 Bayovac strain. The other sequences formed 2 separate monophyletic clades derived from 2 separate subgroups. In more detail, 9 sequences formed a divergent clade within subgroup III. The Italian samples forming this group came from outbreaks in 2013–2015 that had occurred throughout the country. The remaining 12 new sequences were related to subgroups V and VI, creating a new clade tentatively called subgroup VII. Also in this case, the samples came from outbreaks that had occurred around the country, but during an earlier period (2012–2013). The average nucleotide similarity along the G protein gene sequence within each clade was equivalent (98.65% and 98.84% within subgroups III and VII, respectively), suggesting comparable evolutionary behavior. Even if the tree topology based on the less-variable N protein gene sequences (Supplementary Fig. 1) does not allow a clear separation of subgroups V and VI as already reported, 17 the new Italian sequences formed a supported subclade. However, given the small number of sequences, the sequences are included in a more general V–VII clade, following previous topology interpretation. 17 The high similarity among the Italian sequences was maintained; nucleotide identity was 99.74% within subgroup III and 99.58% within the subclade of V–VII groups, respectively.
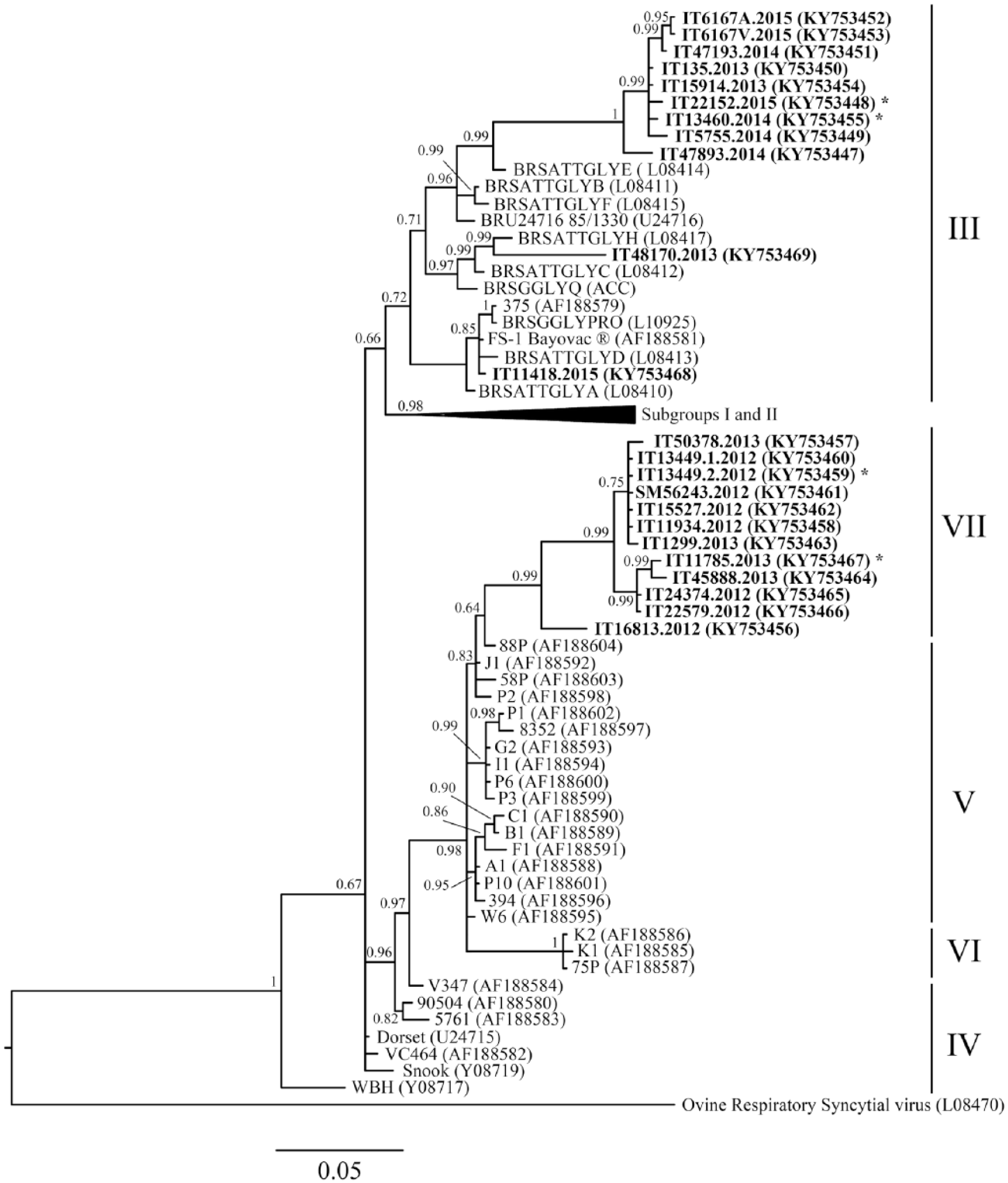
Bayesian tree of bovine respiratory syncytial virus G gene partial sequence. Designations at the ends of the branches refer to the subgroup based on Valarcher et al., 2000. New Italian sequences are reported in bold. The year of collection is indicated as the last part of the sample name. Sequences obtained from animals from vaccinated herds are marked with an asterisk.
Some sequences included in the divergent clade within subgroup III and new subgroup VII came from farms in which vaccination measures were in place (Table 1, marked by an asterisk in Fig.1 and Supplementary Fig. 1), probably because of poor implementation of a vaccine protocol or because of selection pressure from a non-sterile immune response. However, if we consider only the linear immunodominant epitope region along the G protein,8,9,17 the new Italian sequences are quite similar to the ones described previously (Supplementary Table 2). The epitopes were characterized as a region along the G protein crucial for its folding. All of the new Italian sequences had a serine at position 184, the amino acid change that typically differentiates subgroup I from the others. The presence of threonine at position 205 associates the new Italian sequences with subgroups III, IV, and V, as well as the leucine-serine at positions 183-184. The Italian sequences belonging to subgroup III had a mutation from proline to serine at position 194: serine was present at that position only in the samples from subgroups IV and V. The Italian clade forming subgroup VII showed pattern SxSxS at position 190: this pattern was typical of BRSV subgroup V.
Although only a small region of the G protein was analyzed, the similarity between the new Italian sequences and the reference sequences suggests that vaccination could still be useful for animal protection; nonetheless, the genetic and antigenic divergence found in Italy and in several other countries constitutes evidence for BRSV circulation and evolution. Moreover, estimation of the evolutionary rate of G protein coding sequence is in line with the previously published data 17 (4.38 × 10-3 substitutions/site/year, effective sample sizes [ESS] >200), supporting the notion that BRSV evolves after its introduction into a susceptible area and before its extinction as a result of natural immune response or vaccination.
Genetic characterization of the circulating viral strains revealed the presence of at least 3 different variants, demonstrating that BRSV is still evolving. This is particularly important in areas in which vaccination protocols are in place. Viral strains belonging to subgroups V and VI have been identified in vaccinated calves, whereas the vaccine strains belonged to subgroups II (i.e., Rispoval) and III (i.e., Bayovac). 17 Given the genetic and antigenic divergence of those strains, the authors suggested that vaccination does not always prevent infection of calves with BRSV of subgroups V and VI; Italian strains are closely related to both old and recent subgroups. No geographic clustering was evident within the country, probably because of animal trade movements, but the spatial aggregation was maintained when different countries were compared.
The Italian sequences belong to 2 different collection periods; the sequences belonging to new subgroup VII were obtained from samples collected before 2013 and they shared the same evolutionary path with the French and Belgian sequences collected at the end of the 1990s (subgroups V and VI). In contrast, none of the more recently collected samples (2013–2015) belonged to this clade, yet all of them descended from the older strains included in subgroup III. Epitope analysis supports the field data, showing similar amino acid sequences of the 2 Italian subgroups, even though the tree topology shows a clear viral evolution along its branches. This temporal separation supports the active role of herd immunity (natural or by vaccination) in preventing the spread and maintenance of viral infection. Nevertheless, the sequences were all monophyletic and formed a separate clade within subgroup III. The clade topology suggests that, within each subgroup, viral isolates could show a continuum of evolution and that their spread could be limited to very short time periods. Continuous investigation and genetic characterization of positive samples are useful tools for updating our knowledge about BRSV evolution and can inform our understanding of the emergence of new viral strains that may escape vaccination protection.
Footnotes
Acknowledgements
We thank Luigi Trosso for his excellent technical work.
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.