
Abstract
California sea lion adenovirus 1 (CSLAdV-1) has been associated with hepatitis and enteritis in several wild and captive populations of diverse pinniped species. Currently available tests have been limited to pan-adenoviral polymerase chain reaction (PCR) followed by sequencing. We present the development of a quantitative probe-hybridization PCR (qPCR) assay for rapid, sensitive, and specific detection of this virus in California sea lions (Zalophus californianus) and other pinnipeds. This assay did not amplify other mammalian adenoviruses and is able to detect consistently down to 10 viral copies per well. Compared with the gold standard conventional pan-adenovirus PCR/sequencing assay, diagnostic sensitivity and specificity of 100% and 88.2% were found, respectively. The lower diagnostic specificity of this qPCR assay may be the result of the lower limit of detection of this assay compared with the gold standard rather than the result of detection of true false-positives.
Adenoviruses have been detected in wild populations of California sea lions (CSLs; Zalophus californianus) since the late 1970s, associated with viral hepatitis.2,5 Given similarities in virus morphology and clinical presentation of the associated disease in infected dogs and sea lions, it was initially thought that canine adenovirus 1 (CAdV-1) might be the etiology of adenoviral hepatitis in CSLs. In 2011, an agent of viral hepatitis in CSLs (CSLAdV-1) was isolated from 2 wild animals 6 and in fecal samples from 2 animals in an open water–managed collection. 4 In 2013, this virus was detected in multiple species of pinnipeds, including a South American sea lion (Otaria flavescens) and a South African fur seal (Arctocephalus pusillus) in Japan, 10 in South American sea lions in Spain, and in a Hawaiian monk seal (Neomonachus schauinslandi) held in an aquarium in Hawaii. 3
Given that CSLAdV-1 may be the product of a virus jumping from an unknown mammalian endemic host, 4 and that evidence exists of transmission between different otariids and a phocid,3,9 the development of a tool that facilitates fast and specific detection of this virus is essential in understanding its epidemiology and pathogenesis. A quantitative polymerase chain reaction (qPCR) assay has several advantages over conventional PCR as a detection tool, including quantitative information about viral loads, high specificity given specific probe hybridization, high analytic sensitivity, lower expense, and less time required. The objective of our study was to develop a test for CSLAdV-1 with high sensitivity and specificity that allows for quantitative detection of this virus in CSLs and other pinnipeds.
Because of the noninvasiveness of sample acquisition and the prior finding of CSLAdV-1 shedding in feces, we analyzed fecal samples and fecal swabs (n = 191) from CSLs. Samples were obtained between 2006 and 2013. For fecal samples, 50 μg of solid material or up to 400 μL of liquid material were used. For swabs stored in RNA stabilization solution a or phosphate-buffered saline (PBS), 300 μL of liquid material were used. For dry swabs, 500 μL of PBS buffer were added to resuspend the sample, and 300 μL was used for DNA extraction. Additional frozen liver samples were requested from previously sequenced PCR-positive stranded animals. We used up to 50 μg of tissue samples. DNA was extracted using an automated extractor. b
DNA concentrations were measured using a spectrophotometer. c Samples were diluted to 25 ng/μL and analyzed using the CSLAdV-1 qPCR assay. Six additional CSLAdV-1–positive samples from clinical cases were used for assay validation. Results are presented as adenovirus copy number per well.
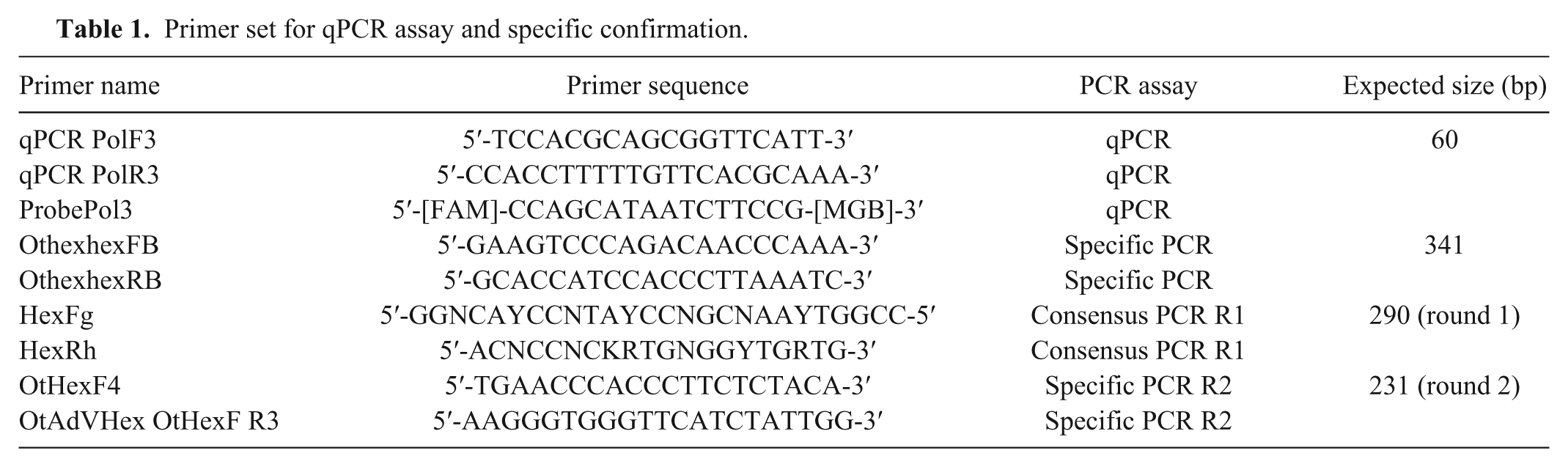
Primers (qPCRPolF3 and qPCRPolR3) and probe (probePol3) were designed to target a conserved area in the DNA-dependent DNA polymerase gene. Primers and probes were designed using commercial software, d with MGB and FAM (Table 1). To obtain template for standard curves, the CSLAdV-1 DNA-dependent DNA polymerase was amplified from a CSLAdV-1–positive sample using a previously published consensus PCR and sequencing assay. 12 This amplicon was 552 bp, and included the sites for binding of the qPCR primers and probe. Dilutions were made with Tris–EDTA buffer and ranged from 107 to 100 copies per 4 μL.
Primer set for qPCR assay and specific confirmation.
Each sample was run in triplicate for CSLAdV-1, with 1 additional well run using universal eukaryote 18s ribosomal (r)RNA gene primers and probe e as an internal control. The master mix for CSLAdV-1 consisted of 20 μL per reaction, with 4 μL of the DNA extract, 10 μL of universal qPCR mix, f 3 μL of water, and 1 μL of primers and probe at concentrations of 3 μM forward primer, 18 μM reverse primer, and 7.5 μM probe. All reactions were run using real-time PCR equipment. g
Samples that did not show amplification of the 18S rRNA gene were discarded. The amplification conditions were 20 s of initial denaturation at 95°C, followed by 45 cycles of 95°C for 3 s, and 62°C for 30 s. The slope, R2, and threshold were calculated with the software incorporated on the thermocycler. g For assay optimization, primer concentrations were adjusted according to a protocol established for qPCR assay optimization. 9
For assay validation, we used 2 approaches to determine the analytic specificity of the assay, in-silico and empirical. For the in-silico evaluation, all known pinniped adenoviruses were aligned to assess homology with the primers and probe. 7 For the empirical evaluation, we ran the CSLAdV-1 qPCR assay against all mastadenovirus samples available in our laboratory, including CLSAdV-2 (GenBank AFS90825.1) and bottlenose dolphin adenovirus 2 (Standorf et al., 2016, pers. comm.).
To evaluate analytic sensitivity of the qPCR, we used standard dilutions (107 to 100 copies/4 μL) of CSLAdV-1 amplicon described previously. Additionally, we analyzed the same dilutions using the previously available nested consensus PCR and sequencing assay for diagnosis of CSLAdV-1 and other adenoviruses. 12 In addition to the analytic specificity and sensitivity, we estimated the diagnostic specificity and sensitivity by comparison of the qPCR assay results to the previously available nested consensus PCR and sequencing assay used for detection of adenoviruses in several species, used here as the gold standard. 12 This nested consensus PCR has been used previously for detection of CSLAdV-1.
In order to calculate diagnostic sensitivity and specificity, a subset of negative samples and all qPCR-positive samples (including positive samples from other pinniped species) were rerun with the gold standard. For additional confirmation of results obtained with the assays, we designed another conventional PCR to amplify a conserved region of the hexon protein using specific primers or a combination of consensus with specific primers (Tables 1, 2). We used a polymerase h for this assay. The initial denaturation was 5 min at 95°C, followed by 45 cycles of denaturation at 95°C for 1 min, annealing at primer 45°C for 30 s, and extension at 72°C for 1 min, followed by a final elongation step at 72°C for 7 min. PCR products were run in 1% agarose gel. Fragments of the expected size were cut and extracted using gel extraction kits. i The gel-extracted PCR products were sequenced in both directions. j
Samples used to calculate diagnostic sensitivity and specificity with qPCR and PCR (specific and consensus primers) for California sea lion adenovirus 1 (CSLAdV-1).*
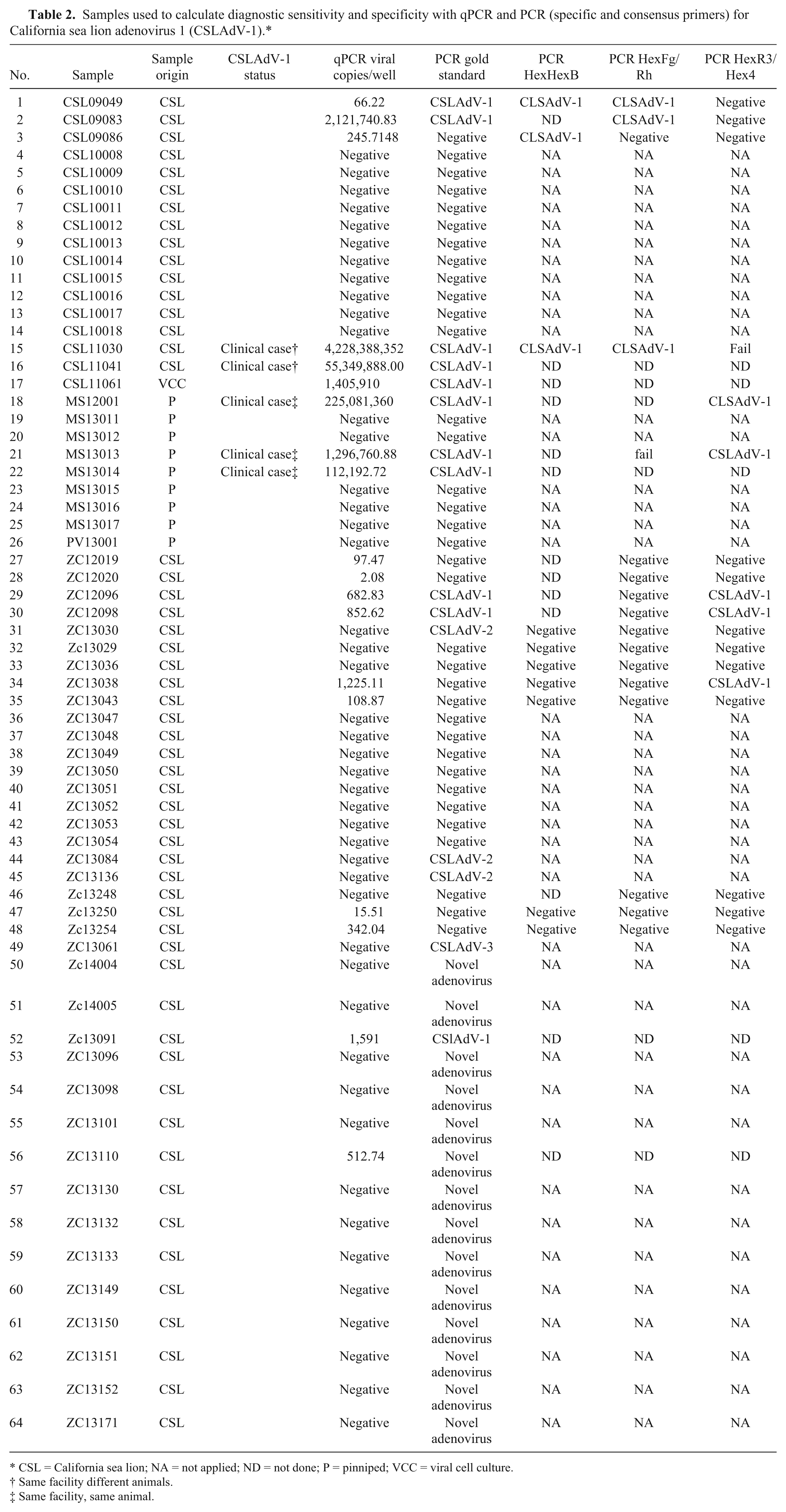
CSL = California sea lion; NA = not applied; ND = not done; P = pinniped; VCC = viral cell culture.
Same facility different animals.
Same facility, same animal.
The linear dynamic range for this assay is 107–101. The diagnostic sensitivity and specificity were calculated from 2 × 2 contingency tables (Table 3). Quantitative PCR–positive animals with DNA sequence 100% homology to existent CSLAdV-1 sequence obtained by gold standard nested consensus PCR/sequencing assay and/or specific PCR/sequencing assays were considered as true positive. We used the Fisher exact test to determine statistical differences of CSLAdV-1 prevalence between wild and open water–managed populations. The standard curve generated represents a linear regression, with an average slope of −3.41 ± 0.05, average efficiency of 96.6 ± 1.86%, and average R2 of 0.99 ± 0.003.
2×2 table used for diagnostic sensitivity and specificity calculations.
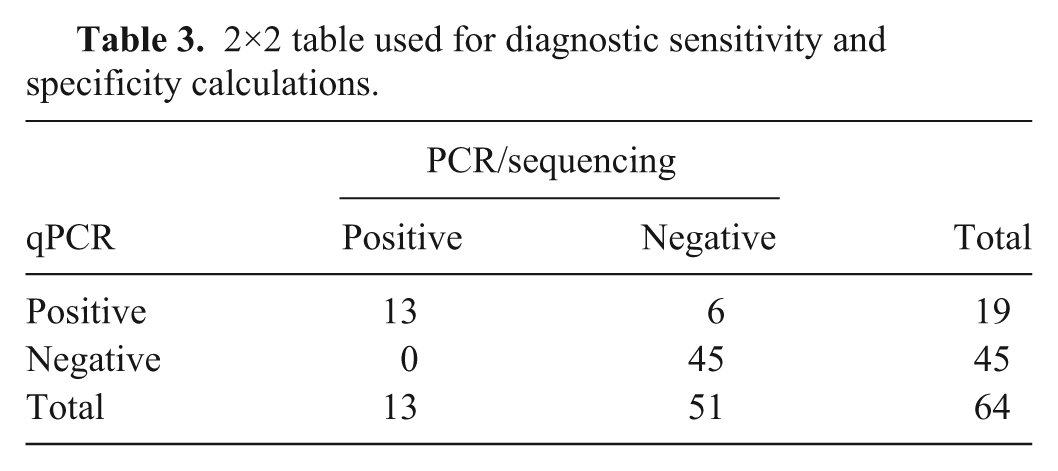
The developed qPCR assay was found to exclusively amplify CSLAdV-1 with no amplification of other mammalian adenoviruses including CSLAdV-2. The analytic sensitivity of this qPCR assay was found to be 10 copies/4 μL. The gold standard (nested consensus PCR/sequencing assay) was able to detect as few as 75 copies/3 µL using pure PCR product. The diagnostic sensitivity of this assay is 100% (confidence interval [CI]: 75.3–100%) and the diagnostic specificity is 88.2% (CI: 76.1–95.6%; Table 3).
Once validated, the qPCR was used for surveillance on the full set of 191 fecal samples analyzed: 29 animals from open water–managed collections and 162 from a rehabilitation center. Thirteen CSLAdV-1 qPCR–positive animals were identified: 3 animals from an open water–managed collection and 10 from wild populations. From those 13 animals, 6 were confirmed by the gold standard nested consensus PCR/sequencing assay (2 from the open-managed population and 4 from wild populations) and 2 additional animals were positive using hexon conventional PCR (Table 2).
The qPCR assay has 88.2% diagnostic specificity compared to the gold standard, hence this assay could generate false positives, especially in samples with low virus loads. However, the additional hexon PCR/sequencing agreed with 2 additional qPCR-positive samples not detected by the gold standard assay. One reason for the low specificity of the qPCR assay may be the higher limit of detection of the gold standard assay. Additionally, the standard curve dilution of the qPCR assay showed a greater analytic sensitivity (10 copies/well) compared to the gold standard assay (75 copies/well), supporting the hypothesis of a lower analytic sensitivity that would affect the diagnostic sensitivity result. Further evaluation of analytic sensitivity of the gold standard could include the use of known negative samples spiked with a known amount of virus particles to evaluate the effect of possible inhibitors in the fecal samples that could affect the analytic sensitivity of the adenovirus gold standard assay.
We found that open water–managed collections (adults plus juveniles) had a prevalence of 7.4% versus 2.5% in wild populations (adults, juveniles, and pups). However, this difference in prevalence was not statistically significant (p > 0.05). The prevalence of CSLAdV-1 found in wild CSLs is similar to a study of CAdVs in the red fox in which 1 of 32 animals had a CAdV-1–positive fecal sample (3.1%) 1 ; in addition, one red fox was positive in liver and kidney, but not feces. In our study, we requested liver samples from CSLs found to be CSLAdV-1 positive in feces; none of the animals was positive for CSLAdV-1. This raises the possibility that, for those animals, the virus had not colonized the liver yet.
One limitation of our study was that, whereas we can detect whether an animal is actively shedding CSLAdV-1, we cannot detect prior exposure. All wild animals positive for CSLAdV-1 were animals involved in an ongoing unusual mortality event or stranded animals. The presence of CSLAdV-1 in these animals is probably related to poor health status. Adenoviruses have been demonstrated to be more virulent in immunocompromised patients and in high-density and/or high-stress situations.8,11
In managed settings, adenovirus may be a health concern in elderly populations of pinnipeds. A CSLAdV-1 outbreak was reported in an aquarium in Japan, in which all infected pinnipeds were >20 y. 9 In addition, CSLAdV-1 was detected in a geriatric Hawaiian monk seal in Hawaii. 3 Further investigation of the role of aging and immune compromise on CSLAdV-1 infection is indicated. Rapid detection of this pathogen is important in zoological facilities, especially in geriatric collections.
Footnotes
Acknowledgements
We thank the staff at The Marine Mammal Center and the Navy Marine Mammal Program for their assistance in sample collection.
Authors’ contributions
G Cortés-Hinojosa and JFX Wellehan Jr contributed to conception and design of the study; contributed to acquisition, analysis, and interpretation of data; critically revised the manuscript; and gave final approval. G Cortés-Hinojosa drafted the manuscript. FMD Gulland and R Rivera contributed to conception of the study; contributed to acquisition of data; and critically revised the manuscript. T Goldstein contributed to conception of the study and critically revised the manuscript. S Venn-Watson contributed to acquisition of data. L Archer contributed to analysis of data. TB Waltzek contributed to design of the study and critically revised the manuscript. G Gray contributed to design of the study. All authors agreed to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
a.
Ambion RNAlater stabilization solution, Thermo Scientific, Waltham, MA.
b.
Maxwell 16 automated extractor, Promega, Madison, WI.
c.
NanoDrop 8000 spectrophotometer, Thermo Scientific, Waltham, MA.
d.
Primer express, Applied Biosystems, Foster City, CA.
e.
VIC probe, Applied Biosystems, Foster City, CA.
f.
TaqMan Fast universal PCR master mix 2×, Applied Biosystems, Foster City, CA.
g.
7500 Fast real-time PCR system, Applied Biosystems, Foster City, CA.
h.
Platinum Taq DNA polymerase, Invitrogen, Carlsbad, CA.
i.
Catalog no. 28706, Qiagen, Valencia, CA.
j.
Life Technologies, Carlsbad, CA.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was funded by research grant N00014-09-1-0252 from the Office of Naval Research to JFX Wellehan Jr.