
Abstract
Bone, despite its relatively inert appearance, is a tissue that is capable of adapting to its environment. Wolff’s law, first described in the 19th century, describes the ability of bone to change structure depending on the mechanical forces applied to it. The mechanostat model extended this principle and suggested that the amount of strain a bone detects depends on bone strength and the amount of muscle force applied to the bone. Experimental studies have found that low-magnitude, high-frequency mechanical loading is considered to be the most effective at increasing bone formation. The osteocyte is considered to be the master regulator of the bone response to mechanical loading. Deformation of bone matrix by mechanical loading is thought to result in interstitial fluid flow within the lacunar–canalicular system, which may activate osteocyte mechanosensors, leading to changes in osteocyte gene expression and ultimately increased bone formation and decreased bone resorption. However, repetitive strain applied to bone can result in microcracks, which may propagate and coalesce, and if not repaired predispose to catastrophic fracture. Osteocytes are a key component in this process, whereby apoptotic osteocytes in an area of microdamage promote targeted remodeling of the damaged bone. If fractures do occur, fracture repair can be divided into 2 types: primary and secondary healing. Secondary fracture repair is the most common and is a multistage process consisting of hematoma formation and acute inflammation, callus formation, and finally remodeling, whereby bone may return to its original form.
Introduction
Bone tissue is highly specialized, and, despite appearing to remain unchanged, is a highly dynamic tissue. It undergoes continual remodeling over an animal’s lifetime, and the ability of bone to respond to injury or increased mechanical demand is unique.
Bone consists of 4 cell types. 31 Osteoblasts produce bone osteoid, a specialized extracellular matrix; osteoclasts are responsible for bone resorption. Bone-lining cells cover endosteal bone surfaces and form a functional barrier with the extracellular fluid. Osteocytes, once thought to be relatively unimportant in bone function, produce key regulatory factors involved in bone formation and bone resorption, and form the basis of a mechanosensation and transduction system. Mechanosensation is the ability of cells to sense or detect strain; mechanotransduction is the process by which these physical forces are transformed into biochemical signals that result in cellular responses by the sensor and effector cells. 72
Bone tissue may change its structure by 2 processes: modeling and remodeling. 129 During growth to adulthood, extensive modeling occurs whereby sequential phases of bone formation and bone resorption alter bone size and shape, under the control of genetic and mechanical factors. Remodeling is the process by which bone tissue is replaced with new bone via the bone multicellular unit (BMU), 130 and is a strictly controlled, coordinated process involving osteocytes, osteoblasts, and osteoclasts. The combination of bone modeling and remodeling allows the bone to adapt to injury and mechanical forces. In this review, we discuss the different mechanisms by which mechanical forces influence bone adaptation, explore bone fatigue and its potential effects, and give an overview of fracture healing.
Mechanical forces and bone adaptation
Wolff’s law and the mechanostat model
Wolff’s eponymous law provides the basis for understanding the effects of mechanical force on bone adaptation. The relationship expressed by Wolff states that “Every change in the form and function of bone or of their function alone is followed by certain definite changes in their internal architecture, and equally definitely alteration in their external configuration, in accordance with mathematical laws.”47,150 The mathematical basis of Wolff’s law has largely been discredited, but the general relationship remains and has been refined by others such as Roux and later Frost.45–47,82,118 At its simplest, the law posits that the mechanical forces applied to bone result in changes to bone structure. This law is illustrated by the increased bone resorption associated with immobilization or microgravity, and increased bone formation seen in response to increased mechanical load.61,101,131
Frost’s mechanostat model takes a multisystem approach to predict the effects of mechanical load on bone adaptation.45,46 The model dictates that the amount of strain a bone detects depends on bone strength (a combination of bone architecture and mass) and the amount of muscle force applied to the bone (the typical peak voluntary mechanical load).45,126 Frost’s model also introduces the concept of minimum effective strain, a genetically determined threshold above which modeling occurs to strengthen bone, and below which remodeling occurs to reduce bone strength.44,46 The actual level of strain at which these changes occur is controversial and currently debated, 117 but the amount of new bone formed appears to correlate with the magnitude and rate of strain. 26
Many of the concepts expressed in the mechanostat model have been demonstrated experimentally. A mouse model of in vivo tibial loading showed that, in response to cyclic compressive load, there was decreased endosteal bone resorption and increased bone deposition beneath the periosteum and endosteum of the cortex. Although, in mouse tail vertebrae subjected to cyclic mechanical loading, this increased bone deposition was the result of an increase in the percentage of bone surface undergoing active bone formation, rather than an increase in the rate of bone formation. 86 Trabecular bone may also realign with peak loading direction. 6 In mice, areas of high local mechanical strain were associated with increased trabecular bone formation, whereas trabecular resorption occurred at sites of low mechanical strain.12,127 Likewise, increased deposition of new bone on existing trabecular surfaces was seen in the metacarpal condyles of 2-y-old Thoroughbred horses subjected to early training. 16 Race training has also been associated with decreased subchondral bone remodeling in the equine distal metacarpus. 70 Similarly, in North American Thoroughbred race horses that run counterclockwise around the race track, this asymmetric loading leads to increased density of the right caudoaxial medial femoral condyle subchondral bone plate and trabeculae. 143 There may also be anatomical differences in the response to mechanical loading given that juvenile sheep that were exercised had increased periosteal bone deposition proximally, but increased osteoclastic remodeling distally. 87
The mechanostat model also takes into account a number of nonmechanical modulators, such as hormones, nutrition, the central and peripheral nervous systems, behavior, drugs, and other environmental factors.45,126 These additional modulators may either positively or negatively affect muscle function, osteoblast and osteoclast function, and the signals detected by the mechanostat, thus altering bone strength and enabling the mechanostat model to explain a number of disease states. 45
All mechanical loads, however, are not created equal. In humans, mouse models, and young horses, low-magnitude, high-frequency dynamic mechanical loads appear to be the most effective at increasing bone formation and bone mineral density.17,68,86,114,117 Muscle contractions provide low-magnitude, high-frequency mechanical loads, as such the effectiveness of this type of load at increasing bone formation is consistent with the mechanostat model, where muscle contraction forces are the main component of the voluntary mechanical load detected by the mechanostat. 45 But bone may become habituated to routine strain. When cyclic loads were applied to the ulna of rats, the longer mechanical load was applied, the less response occurred, 116 and a rest period was required before bone could respond to further mechanical loading.72,123 Thus, cyclical loading with recovery periods is thought to create a more effective bone response, and is therefore considered to be a constructive treatment for improving bone formation.26,72
Age has a marked effect on the response of bone to mechanical loading. Many studies in children have shown that, during growth, bone is highly responsive to loading.11,37,75 For example, a study in humans found that throwing activity when young doubled the strength of the humerus, and one-third of this increased strength remained in adulthood. 147 Similarly, at 12 mo of age, young horses that had been given mild exercise from birth had greater bone area, periosteal circumference, and bone strength in the proximal phalanx and third metacarpal when compared with control horses. 42
In the elderly, however, the response of bone to loading is substantially diminished. Although there may be a reduction in endocortical bone resorption with bone loading in elderly people, more importantly, bone formation under the periosteum is decreased,71,132,140 and bone circumference is a primary determinant of bone strength. 113 Similarly, in aged mice subjected to tibial loading, endocortical bone formation was enhanced but there was no change in periosteal bone formation. 12
Osteocytes and the mechanotransduction network
Historically, osteocytes were thought to have little function in bone; however, these cells are increasingly being recognized as mechanosensors with an essential role in the mechanotransduction network of bone, as well as important roles in mineral metabolism, bone formation, and bone resorption. Osteocytes produce a number of important regulators of osteoblast and osteoclast function. 13 Osteocytes, like osteoblasts, may produce receptor activator of nuclear factor–kappa B (NF-κB) ligand (RANKL), and osteoprotegerin (OPG). 154 RANKL acts as a ligand for the RANK receptor on osteoclast precursor cells and mature osteoclasts. The binding of RANKL to RANK activates a number of signaling pathways that ultimately result in fusion of osteoclast precursors to form multinucleate osteoclasts capable of bone resorption. 15 OPG acts as a decoy receptor that binds to RANKL preventing it from binding to RANK on osteoclasts. 15
Osteocytes also produce sclerostin and dickkopf-1 (DKK1), which are important negative regulators of the Wnt/β-catenin pathway. 13 Sclerostin and DKK1 are antagonists of lipoprotein receptor 5 (LRP5) and prevent Wnt binding and subsequent activation of β-catenin, thereby inhibiting osteoblast activity and bone formation. 90
Fibroblast growth factor 23 (FGF23), a key component in phosphorus homeostasis, is secreted by osteocytes. 14 FGF23 acts on the kidney to increase phosphorus excretion and also inhibits renal 1α-hydroxylase activity leading to decreased production of 1,25-dihydroxyvitamin D (active vitamin D). 64 A number of upstream regulators of FGF23 are also produced by osteocytes, including dentin matrix protein 1 (DMP1), and phosphate-regulating gene with homologies to endopeptidases on the X chromosome (PHEX). 94
Osteocytes are derived from osteoblasts and become entrapped within the mineralized matrix during active bone formation. 43 They are located within lacunae, but have long branching cytoplasmic processes that extend through canaliculi (the lacunar–canalicular system or LCS) and enable them to connect to other osteocytes and cells on the bone surface. 43 Using in vitro and in vivo models, mechanical loading has been shown to result in pressure to, or deformation of, the bone matrix27,114 that creates an interstitial fluid flow within the LCS.112,146 In addition, loading may also induce hydrostatic pressure in the LCS, and cyclic hydrostatic pressure, as generated when walking, has been shown to stimulate bone development in a fetal chick femur model. 66 Although the normal strains placed on whole bone were considered insufficient to stimulate osteocytes in vitro, 155 in vivo models have shown that there is amplification of strain in the LCS because of fluid drag forces on the pericellular matrix. 156 These drag forces are associated with spacing of glycosaminoglycan side chains of proteoglycans and integrin-tethering elements that attach osteocyte processes to the canalicular wall (Fig. 1).62,145,156
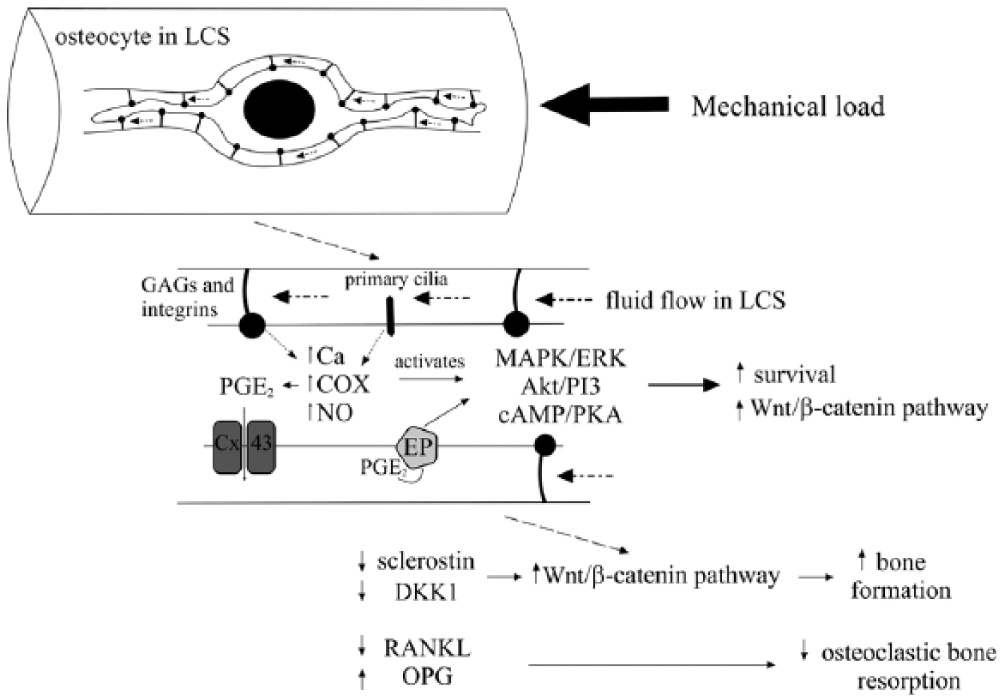
Osteocyte mechanosensation and transduction within the lacunar–canalicular system (LCS). Mechanical load results in interstitial and hydrostatic fluid flow that creates drag forces because of the attachment of osteocyte processes to the canaliculi wall by glycosaminoglycan side chains of proteoglycans (GAGs) and integrin-tethering elements. This may lead to activation of primary cilia and increased prostaglandin E2 production (PGE2), as well as opening of connexin 43 (Cx43) hemichannels releasing PGE2 from the osteocyte. Ultimately, increased intracellular calcium (Ca) and nitric oxide (NO) occur, along with increased cyclooxygenase (COX) activity, leading to activation of cAMP/protein kinase A (PKA), Akt/phosphatidylinositol 3–kinase (PI3K), mitogen-activated protein kinase (MAPK), and extracellular signal-regulated kinase (ERK 1/2) pathways. Binding of PGE2 to PGE2 receptors (EP2/4) also activates cAMP/PKA and Akt/PI3 pathways. Stimulation of these pathways leads to increased osteocyte survival and changes in gene expression, including decreased expression of sclerostin, dickkopf 1, receptor activator of NF-κB ligand (RANKL), and increased expression of osteoprotegerin (OPG). The end result is increased bone formation and decreased osteoclastic resorption in response to increased mechanical load.
The primary mechanosensing mechanism in osteocytes is unclear, but osteocyte ion channels, gap junctions, hemichannels, and primary cilia may all have roles in mechanosensing and mechanotranduction. 27 Gap junctions allow direct communication between osteocytes and osteoblasts. 27 Mechanical load–induced fluid flow increases the expression of the gap junction connexin 43 (Cx43) and induces Cx43 hemichannel opening allowing the release of prostaglandin E2 (PGE2) production.56,77,115 PGE2, together with mechanically induced binding of α5β1 integrins to Cx43, results in activation of the cAMP/protein kinase A (PKA) pathway, phosphatidylinositol 3/kinase (PI3K) pathway, and the PGE2 receptors (EP2/4) preventing osteocyte apoptosis (Fig. 1).9,83 In addition, cell culture models have suggested that interstitial fluid flow may also prevent tissue necrosis factor–α (TNFα)-induced apoptosis of osteocytes. 134
Primary cilia, organelles composed of microtubules that project from nearly all cells, are well-established mechanosensors in the kidney. 128 Hence, there has been interest as to whether primary cilia could have the same function in bone. Although primary cilia are present on osteoblasts and osteocytes, 151 an immunohistochemistry study found that only 4% of osteocytes in the vertebral bone of 6–8-mo-old sheep expressed primary cilia. 30 In addition, a fluid structure interaction model has suggested that the arrangement of the cilia within the LCS was critical in order for them to be stimulated by hydrodynamic pressure. 141 However, an osteocyte cell culture study found that primary cilia appeared to be required for fluid flow–induced PGE2 release from osteocytes (Fig. 1). 91 Therefore, although increasing evidence suggests that primary cilia are involved in bone development, cell differentiation, and proliferation, further experimental evidence is required to determine their role in mechanosensing.104,152
Nevertheless, regardless of the primary sensing mechanism in response to mechanical stimuli, intracellular calcium rapidly increases, 74 increased nitric oxide is released, 5 and cyclooxygenase-2 is upregulated leading to increased PGE2 release.49,115 Ultimately, these changes lead to activation of osteocyte intracellular pathways including mitogen-activated protein kinase (MAPK) and extracellular signal-regulated kinase (ERK 1/2) pathways, cAMP/PKA pathway, Akt/PI3K, and Wnt/β-catenin pathways (Fig. 1).83,109,120,121,135
Mechanical load–induced activation of osteocyte intracellular pathways accordingly results in changes to gene expression, including RANKL, OPG, osteopontin (OPN), DKK1, and sclerostin, 27 all of which are important controllers of either bone formation or bone resorption (Fig. 1). Mechanical loading decreases sclerostin expression in osteocytes, which results in reduced inhibition of the Wnt/β-catenin pathway and therefore increased bone formation; the converse also occurs.87,88,137 Mechanical loading also results in a decrease in the RANKL-to-OPG ratio and therefore inhibition of osteoclast formation and bone resorption.153,157 Consequently, the osteocyte is thought to be the master regulator of bone’s response to mechanical loading.
Bone fatigue
Bone fractures in horses caused by imposition of overwhelming external force (falls or collision with hard objects) are relatively rare. Most fractures are associated with physical activity during which muscles, the weight of the body, and the ground all apply forces to musculoskeletal tissues (muscle, tendon, bone, ligament, meniscus, synovial membrane, joint capsule, and cartilage of various kinds). Even in the least deformable of these (bone), the forces of normal everyday physical activity result in loads applied to, and with consequent deformation (strain) of, bone tissues (cortical or trabecular).
Even these customary strains, which are below the predicted microdamage threshold of the mechanostat model, result in microcracks in both equine cortical and trabecular bone from soon after birth.39,45 Microdamage is removed and repaired by bone turnover (remodeling), a process whereby the skeleton is resorbed and replaced by different cells at a rate that changes with age and physiologic state. In nonathletes, the resistance of bone to deformation is not compromised by microdamage, but, as loads and the number of strain cycles increase, the operational microdamage threshold of bone is exceeded, so that microdamage accumulates, leading to evidence of shear failure at the cement line of the osteon 69 and of collagen, 125 followed by formation of microcracks. Adjacent to fatigue-induced microcracks, osteocytes become apoptotic in areas that later are resorbed by osteoclasts, suggesting that osteocytic apoptosis is closely involved in starting targeted remodeling of the damaged bone. 25 The number of linear microcracks (but not diffuse microdamage) in the cortex is closely related to the subsequent activation of adjacent intracortical bone resorption. 67 Apoptotic osteocytes (staining with caspase-3) and those expressing RANKL and vascular endothelial growth factor (VEGF; associated with osteoclastogenesis) were found to be more numerous in the microdamage region than some hundreds of micrometers away. OPG expression was low near the microdamage, and control nonfatigued tissue expressed OPG. Thus, the neighboring nonapoptotic osteocytes are pro-osteoclastogenic in this targeted remodeling response.
Although resorption spaces within the bone are closely associated with microdamage, 22 the number of microcracks may not be as important as the propensity of cracks to propagate and coalesce into an extending microfracture (although proximal sesamoid midbody fractures in racehorses appear not to be preceded by microcracks). 85
If bone turnover is suppressed, cracks accumulate because they are not repaired by osteoclastic resorption as quickly and can thus grow to a large size, an important factor in influencing clinical fracture risk. 21 Crack extension because of lack of repair may be a critical determinant in catastrophic fracture. 65 Horses trained relatively mildly before ever racing already had less remodeling in comparison to pastured horses, with mild density increases (lower porosity) in the cortex and massive increases in density at fracture disposition sites.40,41 However, compared to untrained horses, continued training is associated with lower third metacarpal bone distal subchondral bone remodeling, 149 less repair of fatigued but high-density bone, and subsequent failure in the form of so-called condylar fracture. 70 Such cracks have been observed previously, 105 and accumulate with increasing age, 139 consistent with higher bone density seen in racing horses >3 y of age when compared with younger racehorses, and the rise in fracture rate after a few years of racing. 106 Exacerbating the propensity for fatigue failure is the rapid bone loss that occurs in the fatigued area, 93 forming large resorption spaces typical of the repair process, which reduces the volume fraction of bone and increases porosity in the predisposition site, thus increasing fracture risk.
The fatigue damage appears to be localized to particular sites in the bone shaft, for instance the proximal humerus, 36 and subarticular sites in the distal limbs. The microdamage is not confined to bone because, in many cases, the first evidence of damage is the response of calcified cartilage in fracture predisposition sites, consisting of thickening, 80 focal retention or hypermineralization,34,38 and increased vascularity. 81 Hyperemia and reduced LCS interstitial fluid flow have been observed in the microdamaged bone shaft, 102 but the signaling responses in the equine epiphysis are unclear.
Fracture repair
Bone fractures may occur as a result of excessive mechanical force applied to normal bone, preexisting microdamage (as discussed above), or normal forces applied to a bone weakened by an underlying disease process (pathologic fracture).31,93 Bone is one of the few tissues that, under the right conditions, can truly undergo regeneration. Furthermore, many of the processes that occur during fracture repair are similar to those that occur during formation of the skeleton. 52 Fracture healing may be divided into 2 types: primary fracture healing and secondary fracture healing.
Primary fracture repair
Primary fracture healing occurs when there is precise reduction of the fracture and the 2 bone ends are directly aligned. The perfect alignment of the fracture ends allows cutting cones to form, osteoclasts resorb bone on either side of the fracture line, and this is followed by bone formation, ultimately leading to reformation of cortical osteons. 107 Despite adequate internal fixation, in practice, there is often a small gap between the fracture ends and this is initially in-filled with woven bone, followed by osteoclastic bone remodeling and reestablishment of osteons, a repair process known as gap healing. 89
Secondary fracture repair
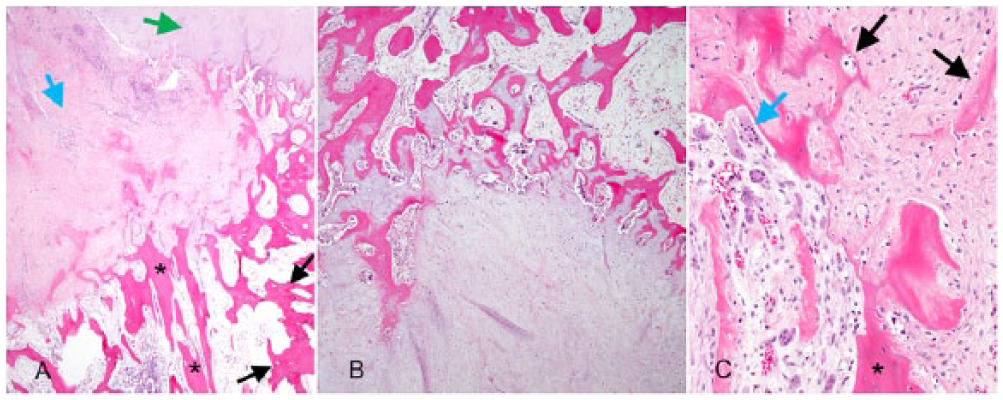
Secondary fracture healing is the most common form of fracture repair and occurs with splinting, casting, intramedullary pins, external skeletal fixators, and other such fixation devices. 89 Secondary fracture repair is typically divided into 3 stages: 1) hematoma formation and acute inflammation; 2) reparative phase with callus formation (Fig. 2A–C); and 3) remodeling and modeling phase, whereby bone may eventually return to its original form. 10 Most experimental data on secondary fracture repair comes from mouse, rat, and rabbit fracture models kept under controlled conditions and so may not be consistent with the reality of fracture repair in practice. Nevertheless, these models do allow us to extrapolate the generalities of fracture repair to other species such as humans and horses.
Dog rib, H&E.
Within the first 24 h of fracture, interleukin-1 and -6 (IL-1, IL-6) and TNFα are secreted by neutrophils and macrophages, and help initiate the repair process by recruiting mesenchymal stem cells.23,35,84 This inflammatory response is essential to the repair process. IL-6 ablation in a mouse fracture model resulted in decreased osteoclastogenesis and a weaker fracture callus, 144 whereas a TNFα knockout mouse fracture model had decreased recruitment of osteoprogenitor cells and impaired intramembranous bone formation. 51
Osteal macrophages (resident bone macrophages) and inflammatory macrophages are important in the fracture repair response because these cells may promote osteoblast differentiation and type 1 collagen secretion. 142 This is clearly illustrated in the fracture callus of macrophage-deficient mouse models that contains less bone and more fibrous tissue.3,142 In addition to producing proinflammatory cytokines, bone and inflammatory macrophages secrete growth factors such as transforming growth factor–β (TGFβ), bone morphogenetic proteins (BMPs), insulin-like growth factor-1 (IGF1), platelet-derived growth factor (PDGF), FGF2, VEGF, and chemokines such as monocyte chemotactic protein-1 (MCP1) and monocyte inflammatory protein-1a (MIP1a), all of which assist with migration of mesenchymal stem cells to the fracture site and stimulate angiogenesis.52,55,122,136 Hence, by 1 wk after the fracture, the initial hematoma is replaced by proliferating mesenchymal cells and granulation tissue (Fig. 2A). 89
In mouse and rat fracture models, osteoprogenitor cells from the periosteum are the primary source of repair cells. 103 However, multipotent mesenchymal stem cells may also come from bone marrow stromal cells, 96 from pericytes around capillaries, 24 from muscle stromal cells, myoblasts, and satellite cells, 32 and from the circulation. 95 Equine mesenchymal stem cells have been shown to be under comparable control systems to those of humans and rodents. 59
Initially, because of low oxygen tension, multipotent mesenchymal stem cells in the center of the fracture site differentiate along the chondrocyte differentiation pathway and produce cartilage. 99 By 7–21 days postfracture in mouse and rat models, the 2 fracture ends are joined forming a cartilage or soft callus. 7 Intramembranous bone formation also occurs at this time, and in mouse and rat models is visible histologically under the periosteum as early as 3 days postfracture, peaking at 7–10 days. 7 As the fracture site becomes more stable and angiogenesis leads to an improved blood supply, mesenchymal stem cells differentiate along the osteoblast differentiation pathway and undergo intramembranous bone formation (Fig. 2C).19,20 From day 14 onward, the cartilage callus is sequentially replaced from outward to inward by woven bone, forming the mechanically stable hard or bony callus (Fig. 2B).7,50,119 In a rat model, at its maximum size on day 14 postfracture, cartilage accounted for 15% of the total tissue and this decreased to 2% by day 35. 50 In comparison, bone tissue made up 75% of the total callus tissue, and this increased to 83% with remodeling of the fracture callus. 50
At the time of peak cartilage formation, OPG expression is high, whereas RANKL and macrophage colony-stimulating factor (M-CSF) expression are low, meaning that osteoclastogenesis is inhibited. 84 However, once bone formation starts, OPG expression decreases, and RANKL and M-CSF expression increase, thus promoting osteoclast formation and mineralized cartilage resorption as part of the process of endochondral ossification. 84 TGFβ and BMP expression are also important during this period of cartilage and bone formation. BMP2 expression appears to be required for maturation and mineralization of the cartilage callus,100,158 and BMP2–8 are involved in osteoblast recruitment and induce differentiation of mesenchymal stem cells into osteoblasts.28,52 Incorporation of BMP2 and BMP6 into the treatment of an equine osteotomy model led to increased bone formation and faster healing at the fracture site. 76 TGFβ2 and 3 are considered to be important in chondrogenesis during fracture repair given that they are highly expressed during this phase.28,52,60,136 TNFα also increases during the bone formation phase where it stimulates osteoblast differentiation. 78
In mouse and rat models, the hard callus undergoes remodeling from as early as 3–4 wk postfracture, a process that continues over months to years.52,92 This phase is actually a combination of remodeling via the BMU, whereby osteoclasts resorb the woven bone followed by formation of mature lamellar bone by osteoblasts, and modeling whereby the callus is reshaped and resized.44,92 IL-1 and TNFα are increased during this phase, and are associated with increased osteoclastic bone resorption.2,52,78,92
Factors that affect fracture healing
The process described above is the ideal, wherein the reduced fracture is stabilized appropriately and there are no underlying disease processes. However, the repair process does not always progress smoothly. Problems with mechanical stability and reestablishment of blood supply, inflammation and infection, nutrition, metabolic bone disease, bone tumors, endocrine disease, and other disease processes may all interfere with healing. Mechanical load, blood supply, and infection are discussed in more detail below. In experimental models, drugs such as fluoroquinolone antibiotics,73,108,138 nonsteroidal anti-inflammatory drugs (NSAIDs), 111 and corticosteroids 148 have been shown to interfere with fracture healing.4,110 The effect of NSAIDs on fracture repair in particular is controversial, and has been reviewed by a number of authors.8,53,54,111
Mechanical load
Experimental models of fracture healing have shown that compression of the fracture site (also known as interfragmentary compression) results in improved fracture repair10,57; however, the timing of load application is important. In a mouse tibial fracture model, compression of the fracture site immediately after fracture inhibited fracture repair regardless of the load applied, perhaps because of damage to new blood vessels. 48 In comparison, a small cyclic load applied 4 days after the fracture resulted in increased strength of the fracture callus, possibly related to the presence of osteoprogenitor cells capable of responding to mechanical load. 48 Hence, it has been suggested that mechanical load is best applied once formation of hard callus has started. 97 The amount of load also affects healing. Absence of strain leads to remodeling and removal of the callus because of disuse atrophy, as could be predicted by Wolff’s law and the mechanostat model. 44 On the other hand, too much mechanical load inhibits healing. 48 Similar to its effects on normal bone, cyclic low-magnitude, high-frequency application of load was associated with faster fracture repair and a stronger callus in a sheep tibial osteotomy model, 58 and micromovement in human tibial fractures treated with external fixation resulted in a shorter time to healing. 79
Blood supply to fracture site
Reestablishing the vascular supply to the fracture site is essential for fracture repair, and impaired angiogenesis is considered a common explanation for delayed fracture healing and nonunion.63,122 For example, the rate of nonunion is higher in humans with open tibial fractures when 1 or more major leg arteries is disrupted. 33 A 2014 study in horses found that the distal aspects of the distal phalanx had increased microvessel density compared with the proximal aspects. The authors suggested that this pattern of microvessel density may correlate with reports suggesting fractures of the distal aspects of the distal phalanx heal better, and the trend is for healing to proceed from distal to proximal in the distal phalanx. 124
Although small amounts of load and movement are considered to improve fracture repair, excessive interfragmentary movement in a sheep osteotomy model resulted in increased deposition of fibrocartilage, decreased bone formation, and decreased numbers of periosteal blood vessels. 29 The timing of blood vessel damage is also important. A rabbit tibia nonunion model showed no difference between union and nonunion groups in blood vessel density at 8 wk, but blood vessel concentration was significantly less at 1 wk postfracture in the nonunion group, suggesting that the effects of poor vascularization are most important early in fracture repair. 18
Infection and inflammation
Although the acute inflammatory response that occurs early in fracture repair is essential for successful healing, excessive inflammation and infection are detrimental. A 2015 study assessing the success of radial fracture repair in horses found that 6 of 8 horses with open fractures did not survive to hospital discharge. 133 Furthermore, the presence of infection at the surgical site was also associated with a decreased likelihood of discharge from hospital. 133 Findings were similar in a study of complete metacarpal fractures in horses, wherein infection of an open fracture was the most common reason for surgery failure and nonunion. 98 Another study showed that internal repairs of equine long bones without postoperative infection were 7.25 times more likely to be discharged from hospital than those with infection. 1 Hence, infection of either surgically repaired or open fractures is an important cause of nonunion and mortality in horses.
Summary
Bone is a highly dynamic tissue that is capable of responding to changes in mechanical load, and this response may be explained by Wolff’s law and the mechanostat model. Low-amplitude, high-frequency loads with rest periods are considered the best conditions to increase bone formation. The response of bone to mechanical load appears to be controlled by osteocytes that detect strains via their cytoplasmic processes within the LCS. However, repetitive strains and increased load when applied to bone may induce microcracks, which, when not repaired, may propagate and lead to catastrophic bone fracture. Normal bone is capable of complete regeneration, whereby the fracture ends are joined by a fracture callus, initially consisting of cartilage and woven bone, but with time replaced by mature lamellar bone. However, a number of factors may interfere with bone healing, particularly infection and impaired vascular supply.
Footnotes
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.