
Abstract
Salmonella enterica subsp. enterica serovar Gallinarum biovar Gallinarum (S. Gallinarum) and biovar Pullorum (S. Pullorum) are 2 poultry pathogens that cause major economic losses to the poultry industry worldwide. Control of both diseases mainly relies on the adoption of biosecurity programs, and success is dependent on accurate and fast detection. Based on this concept, we developed a duplex PCR assay, targeting 2 chromosomal sequences, which allowed us to precisely identify and differentiate S. Gallinarum and S. Pullorum field strains. This assay was validated by testing genomic DNA from 40 S. Gallinarum and 29 S. Pullorum field strains, 87 other Salmonella serovars, and 7 non-Salmonella strains. The serovar identifier region (SIR) primers produced a fragment only in S. Gallinarum and S. Pullorum strains, whereas the fragment from the ratA coding sequence, which was previously demonstrated to differentiate the 2 biovars, was also amplified from other Salmonella serovars. Our results showed that the combination of both SIR and ratA amplifications could be used to identify as well as to differentiate colonies of S. Gallinarum and S. Pullorum reliably. Thus, we believe this methodology can be a useful ancillary tool for routine veterinary diagnostic laboratories by providing rapid, accurate results.
Salmonella enterica subsp. enterica serovar Gallinarum biovar Gallinarum (S. Gallinarum) and biovar Pullorum (S. Pullorum) are the etiologic agents of fowl typhoid and pullorum disease, respectively.2,16 Although S. Gallinarum and S. Pullorum are still referred to as biovars of the same serovar,2,6 genomic analysis indicates that both bacteria have arisen separately from a common progenitor resembling Salmonella enterica subsp. enterica serovar Enteritidis (S. Enteritidis).10,18
Fowl typhoid is a severe acute or chronic disease more common in susceptible mature chickens; pullorum disease is usually a severe acute systemic illness of very young chicks.16,19 In their acute form, fowl typhoid and pullorum disease produce high mortality, 2 although both diseases are characterized by distinct epidemiologies. In fowl typhoid, the bacterium is mainly spread horizontally, whereas in pullorum disease, bacteria can be spread horizontally and vertically, sometimes resulting in long-term persistence in the host.2,4
Fowl typhoid and pullorum disease are responsible for considerable economic losses in the poultry industry worldwide. 2 Between 2010 and 2014, outbreaks of both illnesses were reported to the World Organization for Animal Health (http://goo.gl/4jbcFn), most of them occurring in the developing countries from Central and South Americas, Asia, and Africa although they have also been reported in Europe.
Both S. Gallinarum and S. Pullorum are nonmotile bacteria that are phylogenetically closely related to a lineage of S. Enteritidis.10,11 These 2 biovars are restricted to some avian species, and they share some features such as the somatic antigens (1, 9, 12) in addition to many biochemical traits.6,16 These characteristics make their distinction laborious and dependent on specific differential biochemical characteristics (e.g., dulcitol fermentation and ornithine decarboxylation). Moreover, strains presenting atypical biochemical behavior have also been reported in many parts of the world, making conventional microbiological differentiation for these atypical strains unreliable.5,11,14,15
Several molecular tests, based on the detection of a DNA sequence target, have been proposed in order to identify and/or differentiate field strains of biovar Gallinarum from those of biovar Pullorum.7,8,9,14,15 These tests have the advantage of providing accurate results faster than conventional microbiologic and serologic tests, in addition to properly identifying biochemically atypical strains. However, some of these molecular assays are still laborious and expensive due to the need for additional reagents such as restriction enzymes or they require more than a single reaction.
Previously, we developed a polymerase chain reaction (PCR) assay, based on a polymorphism in the ratA coding sequence (CDS) able to differentiate biovars Gallinarum and Pullorum. 3 However, as reported previously, the ratA CDS may be found in other Salmonella genomes. 3 Thus, misinterpretation might arise if the tested DNA does not come from Salmonella isolates of serovar Gallinarum. In this study, we have overcome this impairment by introducing a second pair of primers into the PCR reaction. These primers target a molecular marker only present in the serovar Gallinarum chromosome.
The rationale for choosing the ratA CDS as a differentiating marker as well as ratA primer sequences have been described previously. 3 A second region, which was found to be specific for serovar Gallinarum, termed ROD9, 18 was chosen to be the “serovar identifier region” (SIR). The conserved nature of SIR has been previously validated in vitro 13 and was also checked using the Basic Local Alignment Search Tool (BLASTn; http://www.ncbi.nlm.nih.gov/blast/Blast.cgi) 1 taking advantage of the newly available Salmonella and non-Salmonella DNA sequences. The forward (5′-TACGGGACGAGTGGGTACTT-3′) and reverse (5′-AGATGCCCCACCACTCAAAG-3′) primers were designed by using Primer-BLAST online software, 20 and their annealing temperature was compatible with that of ratA primers. Primer specificity was first assessed in silico by means of the Primer-BLAST software 20 utilizing the nonredundant database.
Forty S. Gallinarum plus the rough vaccine strain (9R), 29 S. Pullorum, 87 other Salmonella serovars, and 7 non-Salmonella isolates were used to validate this duplex PCR assay (Supplemental Tables 1 and 2; available at http://vdi.sagepub.com/content/by/supplemental-data). Microorganisms were obtained either from national reference centers, namely the Oswaldo Cruz Foundation (Rio de Janeiro, Brazil) and the National Agriculture and Livestock Laboratory (LANAGRO; Campinas, São Paulo, Brazil), or were isolated by our group from poultry examined at the Laboratory of Avian Diseases (Universidade Estadual Paulista, campus Jaboticabal, Brazil). All bacteria utilized herein were identified by serology and biochemical tests at LANAGRO. Among them, 1 strain of S. Gallinarum was unable to ferment dulcitol, and 5 S. Pullorum isolates did not decarboxylate ornithine. Strains were cultivated in lysogeny broth a as described previously. 3 Genomic DNA (gDNA) was extracted using a commercial kit b following the manufacturer’s instructions. The gDNA purity and concentration was checked by spectrophotometry. c American Type Culture Collection strains 9184 and 9120 were used as positive controls for S. Gallinarum and S. Pullorum, respectively.
The duplex PCR assay was set up as follows: 1× buffer with KCl, d 240 μM of each deoxynucleotide triphosphate, d 3.6 mM of MgCl2, d 0.8 μM of each primer, e 1.25 U of the recombinant Taq DNA polymerase, f 20 ng of gDNA template, and ultrapure water e up to 25 μL. The reactions were performed in a thermal cycler g as follows: initial denaturation at 94°C for 3 min, followed by 26 cycles at 94°C for 45 s, 64°C for 45 s, and 72°C for 2 min, with a final step of 72°C for 7 min. The PCR products were analyzed by electrophoresis at 4 V/cm for 60 min in a 1.5% (w/v) agarose gel stained with ethidium bromide e at the concentration of 0.6 µg/mL of gel running buffer. Images were digitized h and analyzed with a commercial software. i
Figure 1 summarizes the main results obtained by applying the duplex PCR. The gDNA from S. Gallinarum strains produced 2 amplicons, namely ratA and SIR, with lengths of 1,047 and 543 base pairs (bp), respectively. Similarly, 2 amplicons measuring 243 (ratA) and 543 bp (SIR) were obtained from amplification of S. Pullorum gDNA samples. The SIR-associated amplicon was not found in any of the other tested strains, although other Salmonella serovars produced the 1,047-bp amplicon (ratA) alone. The gDNA from the commercial rough vaccine strain, S. Gallinarum 9R, was also tested and it produced amplicons similar to those found in the S. Gallinarum field strains. The limit of detection of this PCR assay determined by decimal-diluting the DNA template of both bacteria was 8.5 pg of gDNA.
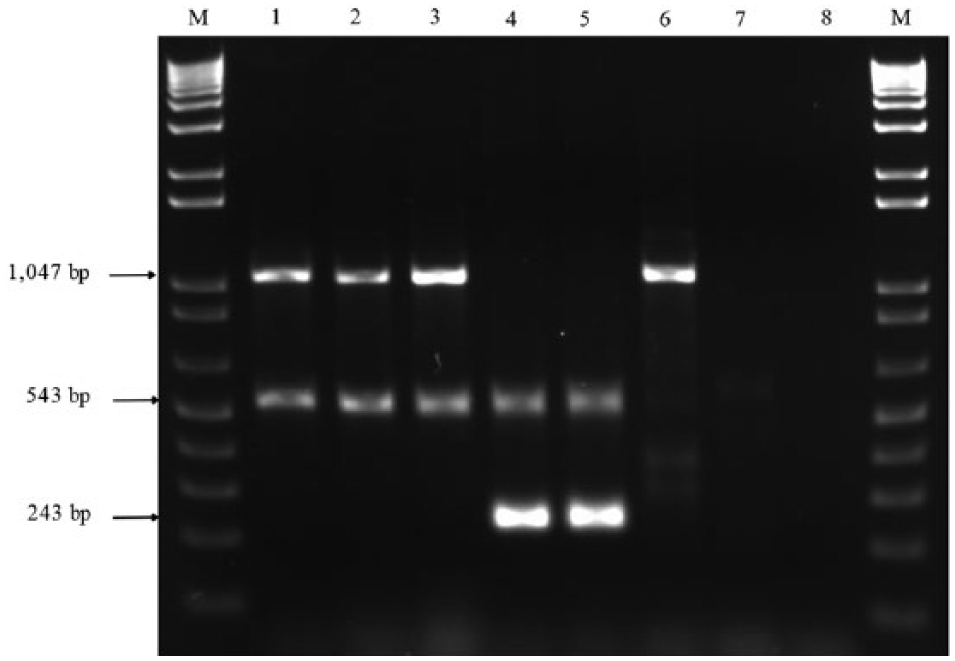
Electrophoresis gel image presenting the combination of fragments obtained after the application of the duplex polymerase chian reaction (PCR) in the strains studied. Lane 1: Salmonella enterica subsp. enterica serovar Gallinarum biovar Gallinarum, strain ATCC 9184; lane 2: S. Gallinarum, strain 287/91; lane 3: S. Gallinarum rough 9R vaccine strain; lane 4: Salmonella enterica subsp. enterica serovar Gallinarum biovar Pullorum, strain ATCC 9120; lane 5: S. Pullorum, strain FCAV198; lane 6: S. enterica subsp. enterica serovar Enteritidis, strain P125109 (PT4); lane 7: Escherichia coli; lane 8: no template control; lanes M: 1-kb DNA ladder. d Note that in lane 6 only the fragment corresponding to ratA gene appears.
Fowl typhoid and pullorum disease are causes of relevant economic losses for the poultry industry in many countries throughout the world. 2 These diseases are present especially where the poultry industry remains in a state of development or in regions where the ambient temperature restricts the opportunity for tight control of the poultry house environment and biosecurity. 16 Therefore, the capability of diagnostic laboratories to provide precise and rapid pathogen identification is a critical component in the implementation of a successful control or eradication plan for both diseases. Conventional microbiologic and serologic methods, however, take 5–7 days to present definitive results, 19 and the existence of biochemically atypical strains5,14 impairs accurate diagnosis and identification.
It has been shown that PCR-based tests are more sensitive and faster for identification of some Salmonella serovars than conventional microbiologic methods.12,17 However, most of the molecular assays have low resolution or still require more than a single step for biovar differentiation.8,9,14,15 The duplex PCR described in our study was able to identify and discriminate all S. Gallinarum and S. Pullorum field strains tested in a single PCR reaction.
The fragment corresponding to SIR (543 bp) was not present in any other Salmonella serovars or non-Salmonella strains, in accordance with a previous report. 13 Thus, the SIR region has been shown to be suitable and reliable as a molecular marker to identify the serovar Gallinarum. As expected, the ratA CDS fragment was observed in other Salmonella serovars (Supplemental Table 2). This situation does not diminish the PCR applicability, as only the combination of ratA and SIR fragments can ensure that bacteria belong either to biovar Gallinarum or to biovar Pullorum.
We believe that the duplex PCR proposed herein could be a powerful tool for veterinary diagnostic laboratories as an ancillary screening test for controlling fowl typhoid and pullorum disease. Analysis of strains from countries other than Brazil would be a valuable adjunct to this study. In addition, we would like to highlight that this duplex PCR was applied only in gDNA extracted from cultured microorganisms and that the efficacy of the assay in terms of detecting and discriminating both bacteria in other type of samples (e.g., clinical samples) requires further investigation.
Footnotes
Acknowledgements
We thank the National Agriculture and Livestock Laboratory (LANAGRO) for providing the S. Gallinarum and S. Pullorum Brazilian strains and LANAGRO and Oswaldo Cruz Foundation for providing the other Salmonella and non-Salmonella strains.
Authors’ contributions
DFA Batista contributed to conception and design of the study; contributed to acquisition, analysis, and interpretation of data; and drafted the manuscript. OC Freitas Neto contributed to conception and design of the study; contributed to analysis and interpretation of data; and drafted the manuscript. AM Almeida contributed to conception of the study; contributed to acquisition of data; and drafted the manuscript. PA Barrow contributed to conception of the study; contributed to analysis and interpretation of data; and drafted the manuscript. FO Barbosa contributed to analysis and interpretation of data; and drafted the manuscript. A Berchieri Jr contributed to design; contributed to analysis and interpretation of data; and drafted the manuscript. All authors critically revised the manuscript; gave final approval; and agreed to be accountable for all aspects of the work in ensuring that questions relating to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
a.
Difco Laboratories Inc., Sparks, MD.
b.
QIAamp DNA mini kit, Qiagen GmbH, Hilden, Germany.
c.
DeNovix Ds 11+ spectrophotometer, DeNovix Inc., Wilmington, DE.
d.
Invitrogen Corp., Carlsbad, CA.
e.
Sigma-Aldrich, St. Louis, MO.
f.
#EP0402, Fermentas, Vilnius, Lithuania.
g.
MyCycler personal thermal cycler, Bio-Rad Laboratories, Hercules, CA.
h.
Gel Doc EZ system device, Bio-Rad Laboratories, Hercules, CA.
i.
Image Lab software, version 4.0.1, Bio-Rad Laboratories, Hercules, CA.
Declaration of conflicting interests
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: The method and primers described in this study were patented at the National Institute of Industrial Property (INPI) under the registration code BR 10 2014 031946-8. Currently, the Universidade Estadual Paulista holds this patent.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was funded by the São Paulo Research Foundation (FAPESP; 2011/23483-1), the Brazilian Federal Agency for the Support and Evaluation of Graduate Education (CAPES), and the National Council of Scientific and Technological Development (CNPq).