
Abstract
Equid herpesvirus 1 (EHV-1) is one of the most economically important equine viral pathogens. Its clinical manifestations in horses vary from acute upper respiratory tract disease, abortion, or neonatal death, to neurological disease termed equine herpesviral myeloencephalopathy, which may lead to paralysis and a fatal outcome. Successful identification of EHV-1 infection in horses depends on a variety of factors such as suitable case selection with emphasis on timing of sample collection, selection of appropriate sample(s) based on the clinical manifestations, application of relevant diagnostic technique(s) and/or test(s), and careful evaluation and interpretation of laboratory results. Several traditional serologic and virus isolation assays have been described; however, these assays have inherent limitations that prevent rapid and reliable detection of EHV-1. The advent of molecular biologic techniques has revolutionized the diagnosis of infectious diseases in humans and animal species. Specifically, polymerase chain reaction (PCR)–based assays have allowed detection of nucleic acid in clinical specimens precisely and rapidly as compared to the traditional methods that detect the agent or antigen, or agent-specific antibodies in serum. The new molecular methods, especially real-time PCR, can be a very useful means of EHV-1 detection and identification. Veterinarians involved in equine practice must be aware of the advantages and disadvantages of various real-time PCR assays, interpretation of viral genetic marker(s), and latency in order to provide the best standard of care for their equine patients.
Keywords
Introduction
Equid herpesvirus-1 (EHV-1; order Herpesvirales; family Herpesviridae, subfamily Alphaherpesvirinae, genus Varicellovirus), which is classified into a subfamily that includes EHV-3 and EHV-4, has a global distribution, and most domesticated horses will become infected or reinfected with this virus during their lives.4,36 EHV-1 can cause acute upper respiratory tract disease, abortion, neonatal death, and neurological disease that may lead to paralysis. 4 Most importantly, EHV-1 establishes lifelong latent infections in the trigeminal ganglia and lymphoreticular tissues associated with the respiratory tract in a high percentage of infected horses.2,4 Reactivation of latent virus can lead to recrudescence of disease and result in viral transmission to in-contact susceptible hosts. The severity of clinical disease associated with EHV-1 infection is influenced by a number of host- and environment-related factors, including but not exclusive of, age, physical condition, immune status, and whether the infection is the result of primary exposure, reinfection, or reactivation of latent virus, as well as secondary bacterial infection of the respiratory tract.2,4,36 Although it appears that all EHV-1 strains can induce abortion in pregnant mares and probably neurologic disease, only certain strains referred to as neuropathogenic or mutant have the potential to cause extensive outbreaks of neurologic disease or equine herpesvirus myeloencephalopathy (EHM) not infrequently associated with a high case-fatality rate (Allen GP, Timoney PJ. Recent advances in our understanding of equine herpesvirus-1 (EHV-1) myeloencephalopathy. In: Proceedings of the 107th Annual Meeting of the United States Animal Health Association. Reno, Nevada, 2007:373–380).40,47 A neuropathogenic phenotype of EHV-1 can result from a single nonsynonymous nucleotide (nt) A to G substitution at position 2254 (A→G2254), leading to a change from neutral asparagine to negatively charged aspartic acid at amino acid position 752 (N→D752).29,40,57 However, there is growing evidence that this is not the only nucleotide substitution responsible for the neuropathogenic phenotype of EHV-1.8,37,43,55 The neurologic form of EHV-1 infection appears to be becoming more prevalent with an increasing number of outbreaks characterized by a high case-fatality rate among affected horses in many countries.8,27,33,37,43,55
Accurate and timely detection of EHV-1 infection in horses is important for equine practitioners, horse owners, breeders, and trainers. The clinical signs of EHV-1–related respiratory disease can mimic those caused by other equine viral and bacterial respiratory pathogens. 42 Similarly, EHV-1–induced neurologic disease must be differentiated from disease caused by other infectious (equine protozoal myeloencephalitis, rabies, arboviral encephalitides, bacterial meningoencephalitis, etc.) and noninfectious (trauma, cervical compression myelopathy [wobbler syndrome], etc.) causes. When confronted with a disease outbreak, confirmation of a provisional clinical diagnosis by means of a rapid, sensitive, and specific laboratory test(s) is essential to ensure that appropriate biosecurity and quarantine measures can be implemented without delay.
Laboratory identification of various clinical forms of EHV-1 infection can be either by direct detection of virus (virus isolation) and/or demonstration of viral antigen or viral nucleic acid detection, or indirectly through serologic evidence of recent infection. The OIE Manual of Diagnostic Tests and Vaccines for Terrestrial Animals recommends any of these methods for the identification of EHV-1 infection. 59 Different diagnostic laboratories will select the test(s) or assay(s) for identification of EHV-1 infection based on available facilities and expertise. However, the clinician or the practitioner needs to be aware of the inherent advantages and limitations of any given test in order to be able to interpret the results of a test appropriately. The primary objective of this article is to review old and new laboratory test procedures available for the identification of EHV-1 infection in horses. These procedures are reviewed based on the type of approach used to achieve identification, with the promising new molecular methods examined in greater detail. This article will also provide an update on advances that have been made in recent years in testing of samples for EHV-1, including cautionary remarks when using molecular tests for its detection in clinical and autopsy specimens.
Specific laboratory identification of EHV-1
Definitive identification of EHV-1 infection can only be made in the laboratory. It is predicated on isolation of the virus, detection of viral antigen(s) or DNA in specimens, or the detection of specific viral antibodies in paired serum samples. Current procedures for the identification of EHV-1 can be broadly classified into several categories, which include the following:
Appropriate sample collection for EHV-1 detection
Direct demonstration of EHV-1
Detection of virus
Virus isolation in cell culture (gold standard) Electron microscopy Detection of viral antigens
Histology combined with immunohistochemistry and fluorescent antibody testing Detection of viral nucleic acid using contemporary molecular biology methods
Nucleic acid amplification–based assays Nucleic acid extraction methods Standard polymerase chain reaction (PCR)–based assays Real-time PCR assays Allelic discrimination real-time PCR assays Insulated isothermal PCR assay Loop-mediated isothermal amplification assay In situ hybridization assay
Indirect demonstration of EHV-1 infection
Serology—detection of antibodies
Complement fixation test Virus neutralization test Enzyme-linked immunosorbent assay (ELISA) based on glycoprotein G Other ELISAs (experimental peptide ELISAs) Agar gel immunodiffusion assay Hematology—analysis of white blood cell counts Cerebrospinal fluid analysis
Detection of EHV-1 during latent infection
I. Appropriate sample collection for EHV-1 detection
It is imperative to collect specific clinical sample(s) at the appropriate time by selecting the most suitable case(s) within an affected group of horses (Figs. 1, 2).28,41 During outbreaks, it is important to collect samples from in-contact horses that are febrile, that may not show any other clinical signs at the time but which may develop clinical signs later, or that could be asymptomatic cases of the infection. It is especially important that nasopharyngeal and/or nasal swabs be collected as early in the course of disease as possible; this is especially important in suspected cases of EHM in which neurological signs appear toward the end of the viremic phase of infection by which time virus shedding from the respiratory tract is waning or may have ceased. Virus shedding from the respiratory tract is generally short lived (<10 days post-infection [dpi]), may be intermittent, and is most reliable during the acute early, febrile phase of respiratory infection (1–5 dpi).25,26,28 The amount of virus shed in nasal secretions tends to correlate with an increase in body temperature. 41 Nasal and/or nasopharyngeal swab samples collected 10 days or later after the onset of clinical signs are less likely to yield positive results on attempted virus isolation (VI). Nasopharyngeal swabs (40 cm in length) are preferred over regular nasal swabs. 45 The swabs should be removed, transferred to viral transport medium containing antibiotics (preferred) or, at the very least, normal saline for testing, transported overnight at 4°C to the laboratory. Furthermore, during the viremic phase of the disease (4–10 dpi), before the appearance of neurologic signs, virus also can be detected in peripheral blood mononuclear cells (PBMCs [buffy coat fraction]). Approximately 10–20 mL of blood should be collected aseptically into ethylenediamine tetra-acetic acid (EDTA; the preferred anticoagulant). On occasion, the virus can also be detected in the cerebrospinal fluid (CSF) in conjunction with the appearance of neurological signs (7–16 dpi).25,26,28 However, while brain and spinal cord (lumbar region) samples collected at autopsy are not the most reliable for VI, such samples are useful for confirmation of viral DNA by PCR and demonstration of characteristic microscopic lesions associated with EHM. The placenta, lung, liver, spleen, and thymus should be collected aseptically in suspected cases of EHV-1 abortion or death in neonatal foals for virus detection. Portions of these tissues can also be collected in 10% buffered formalin along with spinal cord and brain from suspect cases of EHM for histologic (hematoxylin and eosin) and immunohistochemical (IHC) examinations. 15 Samples also can be collected in glutaraldehyde for electron microscopic examination.
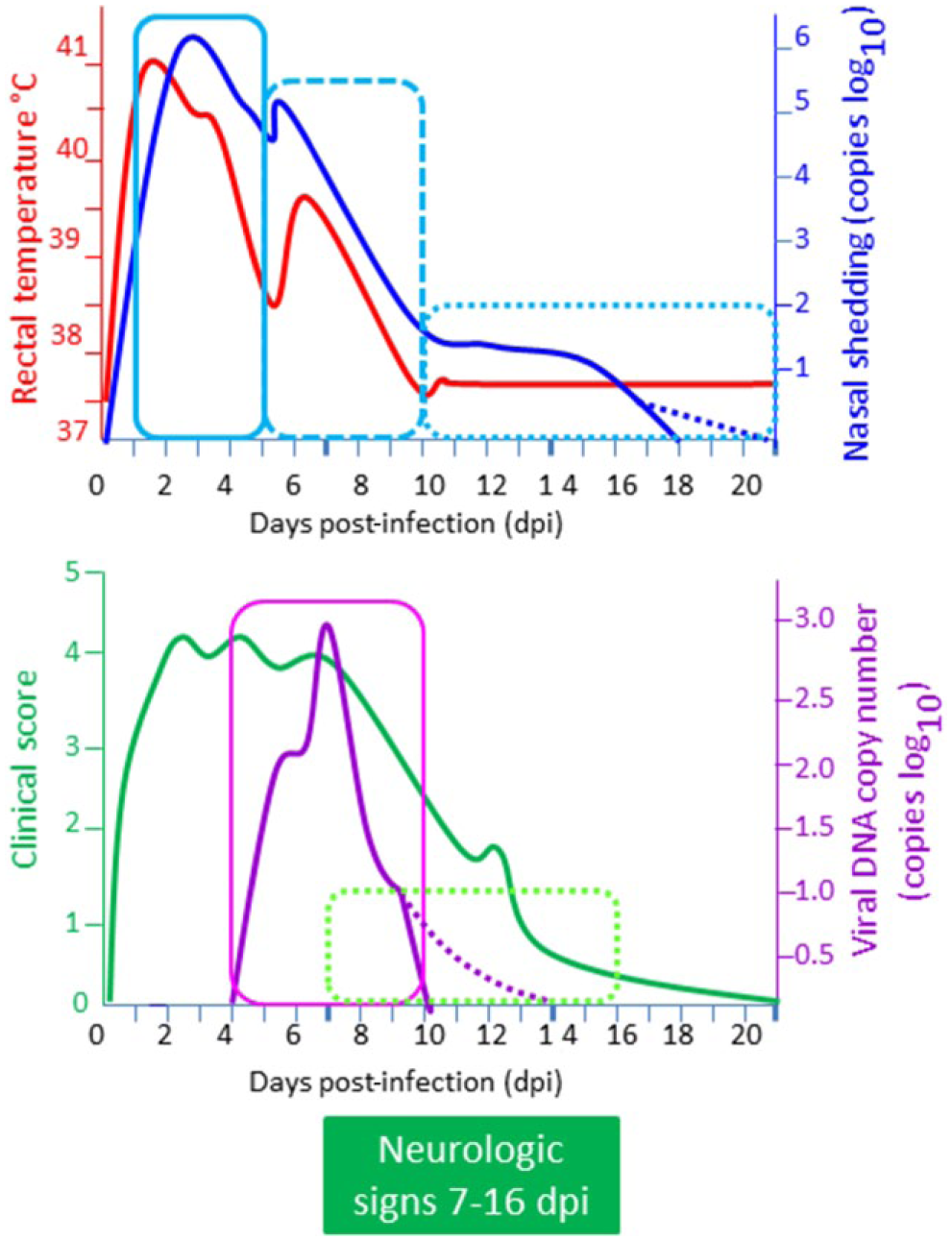
Case selection and appropriate timing of collection of various clinical samples for laboratory identification of Equid herpesvirus 1 (EHV-1) infection.
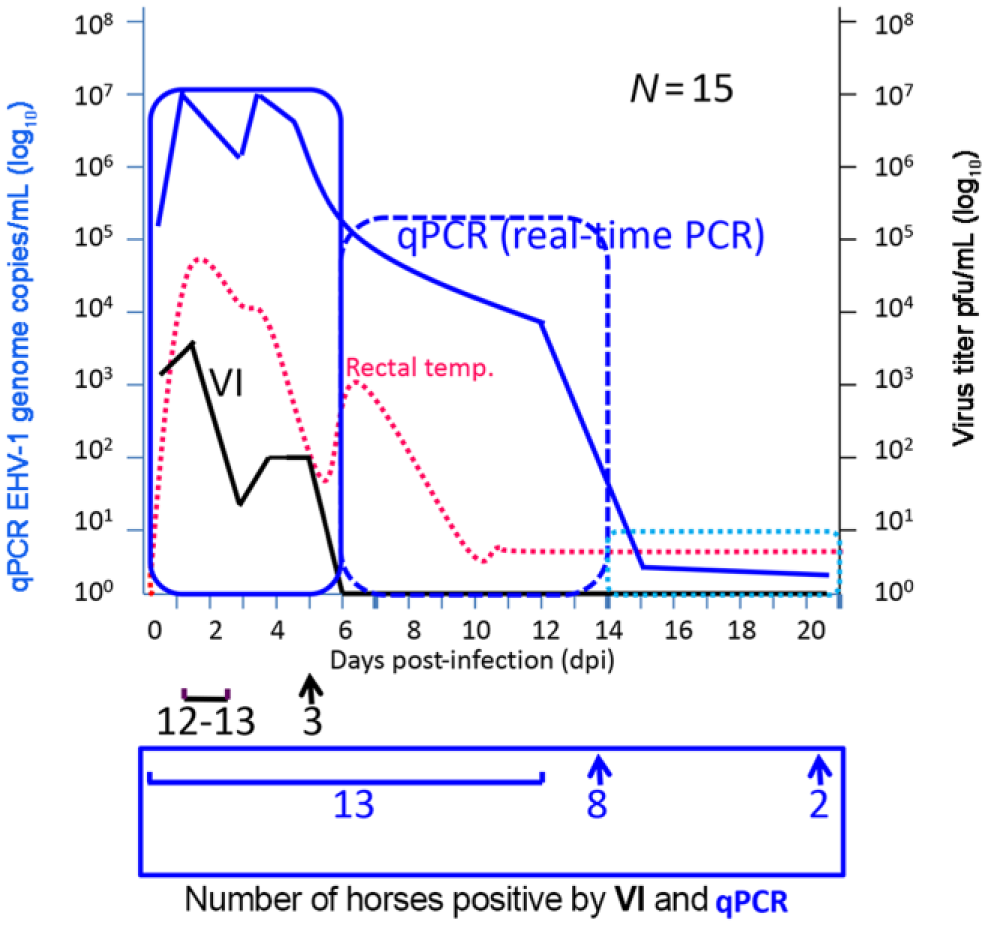
Direct comparison between virus isolation (VI) and real-time polymerase chain reaction (PCR) using nasal swab samples from horses (n = 15) experimentally inoculated with Equid herpesvirus 1 (EHV-1). Days post-infection (dpi) depicted on the X-axis. EHV-1 genome copy number (copies/mL in log10) present in nasal swab samples is depicted in blue on the left Y-axis. Infectious EHV-1 particles in nasal swab samples are depicted in black on the right axis. Rectal temperatures of the horses are depicted by red dotted lines. Attempted VI in cell culture only detects EHV-1 ≤5 dpi. In contrast, quantitative real-time PCR (qPCR) could detect EHV-1 DNA in nasal swabs at least ≤21 dpi (solid blue line). The dpi where both infectious live virus and viral DNA can be detected is shown in vertical solid blue box. During 1–6 dpi, 105–107 EHV-1 genome copies/mL can be detected. Then, the DNA copy number drops rapidly to 14 dpi (vertical broken line blue box). By 21 dpi, the virus copy number is reduced to 1–10 EHV-1 genome copies/mL (horizontal dotted light blue box). At the bottom of the graph is shown the significantly reduced number of horses shedding live EHV-1 by 5 dpi (12–13 horses were positive at 1–3 dpi and only 3 horses [3/15] are positive at 5 dpi by VI), whereas EHV-1 DNA can be detected in a significant number of horses (13/15) up to 12 dpi. Subsequently, the number of horses positive by qPCR is significantly reduced after 12 dpi. Modified from Perkin et al. (2008). 44
All samples submitted for laboratory testing should be accompanied by completed sample submission forms. Information provided should include complete history, differential diagnosis, and type of samples submitted for laboratory examination. All samples for VI and demonstration of viral antigens and/or nucleic acid should be stored and transported at 4°C. It is very important not to freeze unclotted whole blood samples submitted for VI. Following collection, samples should be shipped via overnight delivery to a laboratory qualified to undertake the testing.
II. Direct demonstration of EHV-1
A. Detection of virus
1. Virus isolation in cell culture (gold standard)
Virus isolation unequivocally demonstrates the presence of infectious virus in a sample and, as a result, is regarded as the “gold standard” for laboratory identification of EHV-1 infection. 59 EHV-1 isolation can be attempted from nasopharyngeal and nasal swabs, bronchoalveolar lavage fluid, tissues of aborted fetuses (placenta, lung, liver, thymus, spleen), buffy coat cells (PBMCs), and central nervous system material from cases of neurologic disease in continuous cell lines (rabbit kidney-13 [RK-13], equine dermis [ED], and baby hamster kidney-21 [BHK-21], Madin–Darby bovine kidney [MDBK], pig kidney-15 [PK-15]) and primary or diploid equine cells (equine lung or kidney cells and equine endothelial cells). 59 Unlike EHV-4, which requires equine-derived cells for isolation, EHV-1 can be isolated in a variety of non–equine-derived cell types. However, the sensitivity of different cells and cell lines can vary significantly, which can result in false-negative results (Table 1).
Outcome of attempted virus isolation from clinical specimens of equine herpesviruses (EHV-1, -2, -4, and -5) in various mammalian cell lines.*
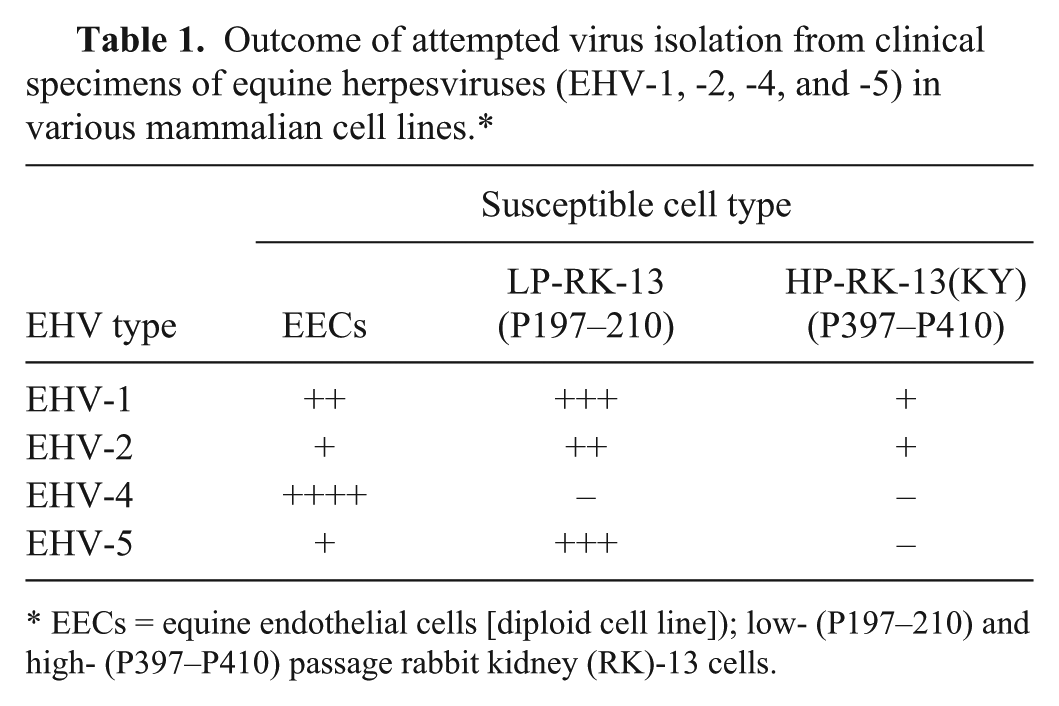
EECs = equine endothelial cells [diploid cell line]); low- (P197–210) and high- (P397–P410) passage rabbit kidney (RK)-13 cells.
Tissue samples for VI should preferably be kept at 4°C, and specifically not −20°C, until inoculated into cell culture. If these samples cannot be processed for laboratory testing within 24 hr after collection, they should be stored at −80°C. However, storage of whole blood samples or buffy coat cells at −80°C will reduce the chance of isolating the virus in cell culture. 4 Therefore, blood samples collected for buffy coat separation should not be frozen. Following inoculation into susceptible cell lines, EHV-1 produces a characteristic cytopathic effect (CPE) in 5–7 days. The outcome of attempted VI depends on the cell line(s) used. The identity of virus isolates recovered from clinical specimens must be confirmed by PCR, indirect fluorescent antibody testing (IFAT), or neutralization assays using EHV-1–specific antisera or monoclonal antibodies. VI can be a challenge for laboratory personnel because propagation of EHV-1 in cell culture is a more labor-intensive and time-consuming procedure, and can be subject to contamination. Accordingly, PCR has largely superseded VI in most laboratories that test specimens for EHV-1.
2. Electron microscopy
Electron microscopy has been used to demonstrate EHV-1 in tissue culture fluid harvested from laboratory cell cultures and in autopsy material. Electron microscopy can be combined with histologic examination of autopsy tissues.
B. Detection of viral antigens
1. Histology combined with immunohistochemistry and fluorescent antibody testing
Characteristic pathological changes can be demonstrated in tissues from aborted fetuses and in spinal cord samples collected from autopsied horses with EHM on histological examination. These changes include the presence of characteristic Cowdry type A intranuclear eosinophilic inclusion bodies in liver cells and respiratory tract epithelium of aborted fetuses, as well as vasculitis and thrombosis in small blood vessels of the spinal cord. IHC staining methods to detect viral antigen in paraffin-embedded tissues of aborted equine fetuses or neurologic cases have been described.14,15,23 IHC staining along with hematoxylin and eosin examination is particularly useful for the simultaneous evaluation of lesions and identification of the infectious agent in infected tissues. Similarly, FAT can be used to demonstrate viral antigen in frozen tissue sections. Frozen sections of postmortem material can be stained with conjugated or unconjugated monoclonal antibody or polyclonal antiserum to EHV-1. 59 Neither FAT nor IHC can discriminate between neuropathogenic and non-neuropathogenic strains of EHV-1. Fluorescein isothiocyanate (FITC)-conjugated polyclonal swine antiserum to EHV-1 can be obtained from the National Veterinary Services Laboratories of the U.S. Department of Agriculture, Ames, Iowa. Furthermore, both assays can be performed on infected cell monolayers, which can be used to confirm the identity of EHV-1. Both FAT and IHC testing must include positive and negative controls consisting of sections of known EHV-1–infected and uninfected tissues.
C. Detection of viral nucleic acid using contemporary molecular biologic methods
1. Nucleic acid amplification–based assays
Several molecular techniques based on PCR have been developed to detect viral nucleic acid in clinical specimens; such techniques have proven very useful in the diagnosis of many animal and human diseases. These laboratory techniques are highly sensitive, specific, and rapid. They have gradually replaced traditional methods of VI and demonstration of viral antigens in clinical specimens.9,19,22,30,58 The PCR-based techniques do not require the presence of infectious virus in the clinical samples. These techniques can detect EHV-1 nucleic acid in clinical specimens, tissue culture fluid from inoculated cultures, and paraffin-embedded tissues.
Polymerase chain reaction assays (standard, nested, and real-time) target a number of EHV-1 genes (gB, gC, gD, and DNA polymerase), but because some of these genes are highly variable among EHV-1 field isolates, consequently not all primers and probes have similar or equivalent sensitivity. To avoid false-negative test results, the primers and probes should be designed based on highly conserved regions of these genes, and assays should be properly developed and validated based on testing a significant number of well-characterized clinical samples before they are recommended for routine use.
Other factors affecting nucleic acid amplification–based assays
Presence of PCR inhibitors in the reaction tube. Hemoglobin and lactoferrin have been identified as PCR inhibitor components of erythrocytes and leukocytes, respectively. In addition, some anticoagulants (e.g., heparin) interfere with the PCR assay. These PCR inhibitors act primarily by inactivating the Taq DNA polymerase used in PCR assays. Various protocols and DNA extraction procedures are available to purify DNA and eliminate these inhibitors prior to performing the assay. However, these extra steps are time-consuming, may not completely remove inhibitors, or may lead to a loss of target DNA resulting in decreased sensitivity of the real-time PCR assay. Consequently, the detection of EHV-1 DNA in blood or buffy coat cells may be less sensitive and result in significant variation in sensitivity of PCR assays among laboratories. Incorporation of internal controls in the PCR reaction will enable identification of PCR inhibitors in the reaction tube. The internal controls either come with PCR reagent kits or they can be purchased. They also can be designed in-house and commercially synthesized, but should be tested and proven not to interfere with the PCR amplification reaction.
Nucleic acid degradation. All samples should be collected and kept cold (4°C) until delivered to the laboratory for processing. It is recommended to store the extracted nucleic acids (DNA or RNA) at −20°C (short term) or −70°C (long term) until further use. Care should be exercised every time a sample is taken out of storage for analysis. Repeated freeze–thaw cycles will degrade the nucleic acid and, therefore, it is recommended that the eluted nucleic acid (DNA or RNA) be aliquoted into multiple tubes for 1-time use. However, this is a labor-intensive task.
Nucleic acid extraction method. The method used for nucleic acid extraction influences the sensitivity of molecular assays.
2. Nucleic acid extraction methods
The use of molecular assays for the detection of EHV-1 and other viruses in clinical specimens requires stringent nucleic acid extraction methods to ensure the stability of nucleic acids.16–18 Commercial reagent kits use glass fiber filters, silica gel spin columns, and magnetic beads to trap DNA present in clinical specimens.2,27–29 The availability of new commercial reagent kits has increased the yield, purity, and quality of extracted viral nucleic acids by removing contaminating genomic DNA, other RNAs, and reverse transcription (RT)-PCR enzyme inhibitors present in clinical specimens. To overcome the inherent problems associated with glass fiber filters and with silica gel spin column–based nucleic acid purification systems, a method has been developed that uses magnetic microsphere beads (0.5–10 µm diameter) to selectively bind nucleic acids.7,20 In general, a microspheric bead–based approach results in more consistent nucleic acid recovery because beads can be fully resuspended in solution to enable more thorough mixing, washing, and elution.
Various veterinary diagnostic laboratories around the world continue to use reagent kits based on fluid phase, solid phase, or magnetic separation techniques for EHV-1 DNA isolation and purification from various specimens (e.g., nasopharyngeal and nasal swabs, and tissues). There have not been many studies that directly compare the efficiency of EHV-1 DNA extraction using various kits based on these 3 different nucleic acid extraction techniques. Interestingly, magnetic beads bind both viral DNA and RNA, and nucleic acid purified with this method could be used for detection of either DNA or RNA viruses.
Manual versus automatic extraction of nucleic acids
Nucleic acid isolation from clinical specimens is a tedious process, and often bottlenecks when testing large numbers of specimens. Most of the first-generation commercial nucleic acid extraction kits described above were designed for manual extraction of viral nucleic acid from clinical specimens. There are numerous disadvantages to manual extraction methods, including limited-throughput, labor-intensiveness, the need for specially trained staff to perform extraction, and technician-dependent variability in the efficacy of extraction. The samples must be manipulated multiple times, which can increase the risk of cross-contamination between samples. In addition, some manual extraction kits use ethanol to precipitate the nucleic acids, and if not properly removed, excess ethanol residues can inhibit the PCR reaction downstream. The advent of automated instruments to extract nucleic acids from clinical specimens has helped to overcome some of the shortcomings posed by manual extraction. Studies comparing manual and automated extraction methods have reported automated extraction to be equivalent and, in some instances, superior to manual methods.20,32 Automated extraction instruments are manufactured by a number of different companies, and the instruments vary in specimen capacity per run and cost per sample for processing. Most companies offer their own reagent kits to be used with their instruments.
Automated extraction systems have inherent advantages over manual methods. These systems keep sample manipulation to a minimum, thus reducing the risk of cross-contamination of the samples caused by human error. Many of the instruments are closed systems, further reducing the risk for contamination. Recovery of nucleic acid from an automated instrument is consistent and reproducible. These instruments are designed for “walk-away” operation and, therefore, are less labor intensive than manual extraction. Once these systems have been installed, validated, and proper maintenance procedures are in place, quality control is less expensive than that required for manual extraction. However, there are several potential drawbacks to automated extraction systems. The initial cost of the instrument is significant, and the instruments are most economical when used to their full capacity (i.e., fully loaded with samples). Therefore, a significant number of samples may need to be processed in order to justify the capital investment in the purchase or lease of these automated instruments. In addition to the cost of the instrument, the cost of reagents and disposables may be high and this cost needs to be considered. Some instruments have large footprints and require space that may not be currently available in a laboratory. Some companies are now manufacturing smaller models that handle fewer samples (e.g., 24–48 samples). These smaller machines are less expensive and may be a viable option for smaller laboratories that process fewer specimens.
3. Standard polymerase chain reaction (PCR)-based assays
A variety of type-specific PCR primers and probes can distinguish between EHV-1 and EHV-4, as well as other EHVs (EHV-2, EHV-3, and EHV-5). A multiplex PCR assay for simultaneous detection of both EHV-1 and EHV-4 nucleic acids has been described. The OIE manual describes a more sensitive nested PCR assay targeting the glycoprotein B genes (gB) of EHV-1 and EHV-4, which allows identification and discrimination of these 2 EHVs. 59 Nested PCR assays are prone to give a higher number of false-positive results because of sample carryover and cross-contamination, and their use should be avoided in the routine laboratory detection of EHV-1.
4. Real-time PCR assays
A number of real-time PCR assays targeting various EHV-1 genes (gB, gD, and viral DNA polymerase [open reading frame 30, ORF30]) have been described in the literature (Allen GP. New insight into equine herpesvirus-1 (EHV-1) neurological disease. In: Equine Disease Quarterly. Department of Veterinary Science, College of Agriculture, University of Kentucky: Lloyd’s, 2006:2).1–3,16,17,30,35,41,44,46,52 However, very few of these assays are properly designed and validated. Real-time PCR assays targeting gB and gD are used for measuring the viral copy number (“viral load”) in samples by some laboratories. 46 Two methods are used to quantify the viral DNA copy number in clinical specimens: relative quantification using cellular housekeeping genes (e.g., β-actin), or absolute quantification using a standard curve based on plasmid dilutions of the target gene (Fig. 3). However, quantification of viral DNA copy number in a clinical sample is challenging and prone to experimental error for the following reasons:
The sample collection method is not uniform given that the amounts of mucus, nasal discharge, debris, and epithelial cells can vary between specimens. Also, the method of swab collection (swab length, variation in contact time with the mucosa [2–20 sec], together with type and volume of transport medium [0.1–7 mL]) can vary.
Nasopharyngeal and nasal swabs are a source of very few epithelial cells and leukocytes. Thus, cellular genes should not be used for correcting DNA purification efficacy and calculating the viral DNA copy number in nasal secretions. Absolute quantification based on a standard curve should be used to quantitate virus in nasal secretions; this method will provide a more accurate assessment of viral DNA copy number in specimens.
Quantification of viral DNA can be performed using a plasmid standard curve that will allow counting the exact viral DNA copy number or using 10-fold serial dilutions of tissue culture fluid containing a known virus titer (plaque-forming units/mL). Both approaches have advantages and disadvantages. Plasmid DNA prepared from Escherichia coli is often contaminated with RNA, which increases the A260 measurement and inflates the copy number calculation used to prepare the standard curve. On the other hand, 10-fold serial dilutions will have better extraction comparability, but they contain both non-infectious and infectious particles and therefore the quantification, which is based on infectious particle only, is also not accurate. In addition to this, different laboratories use different standards for quantification, and it is important to note that viral particle counts cannot be compared if they were not run in the same laboratory and against the very same standard.
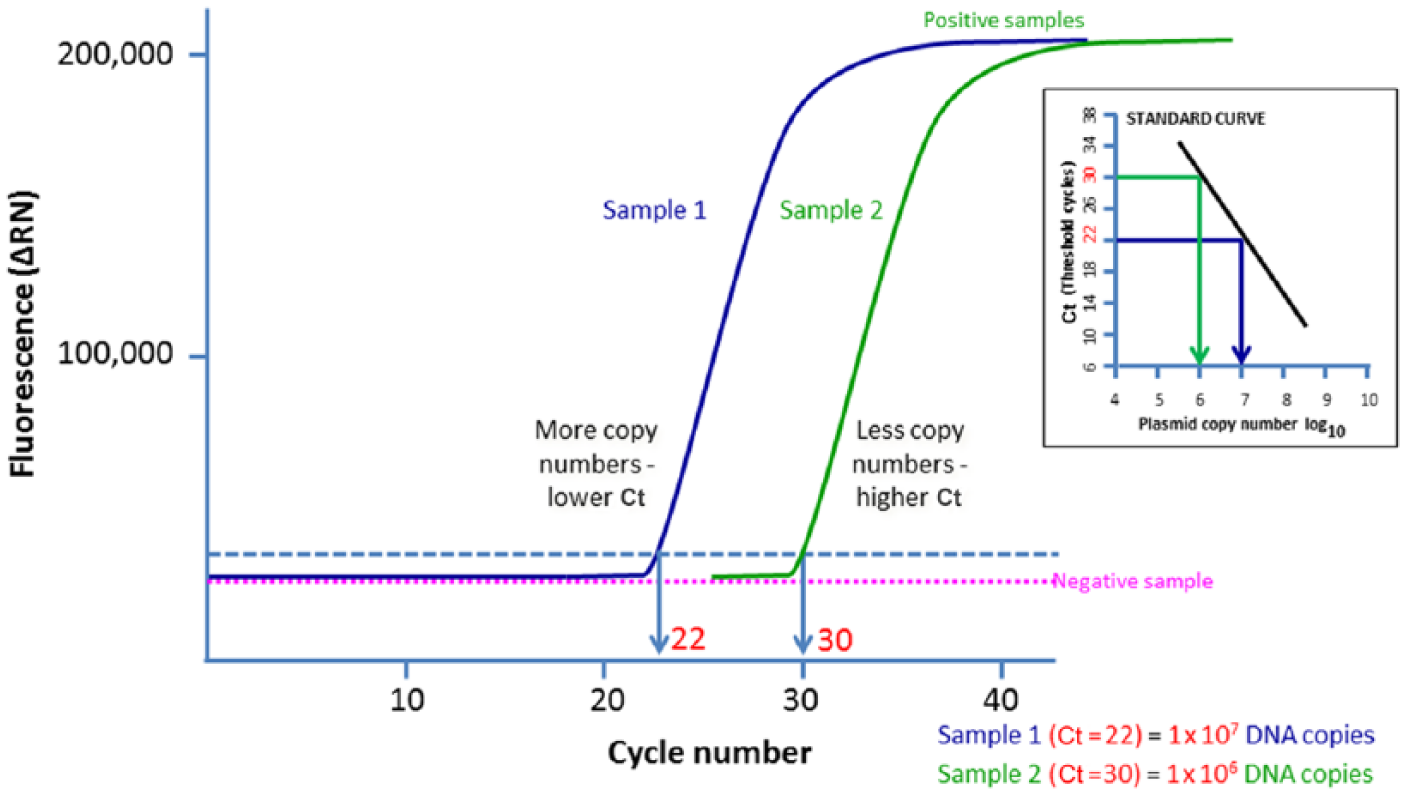
An example of qPCR assay based on absolute quantification. The more copy numbers of Equid herpesvirus 1 (EHV-1) DNA present, the sooner an increase in fluorescence is detected. Based on the standard curve, the threshold cycle (Ct) value can be converted to DNA copy number (e.g., Ct value of 22 = 1 × 107 DNA copies, or Ct value of 30 = 1 × 106 DNA copies).
5. Allelic discrimination real-time PCR assays
Identification of the single nucleotide polymorphism in ORF30 (A→G2254) led to the development of an allelic discrimination real-time PCR assay to distinguish between potential neuropathogenic and nonneuropathogenic EHV-1 strains (Allen GP, 2006, New insight into equine herpesvirus-1 (EHV-1) neurological disease). The first allelic discrimination real-time PCR assay, described in 2007, had a distinct advantage over existing PCR assays in that it could simultaneously detect and genotype EHV-1 strains (Fig. 4; Allen GP, 2006). However, subsequent evaluation of clinical samples using this assay in several laboratories demonstrated that the assay lacks adequate sensitivity for routine application and may also generate false dual-positive (A2254+G2254) results, seriously compromising its usefulness for A2254/G2254 genotype differentiation. Additionally, false-negative results are produced in this assay by the presence of a single, additional nucleotide substitution within ORF30, at position 2258. 51 A new allelic discrimination EHV-1 real-time PCR assay targeting ORF30 has been developed, and its sensitivity and specificity has been compared with the original assay described in 2007 (Allen GP, 2006). 31 The new assay has the following advantages over the original assay:
The assay has been validated with a large number of archived tissue culture fluid samples.
The assay was 10 times more sensitive than the original assay, with a lower detection limit of 10 infectious virus particles.
The new assay was able to accurately discriminate between A2254 and G2254 genotypes, whereas the previous assay produced 20 false dual-positive results with only 1 actual mixed A2254+G2254 genotype confirmed.
The assay offers greater sensitivity and accuracy for the detection and A/G2254 genotyping of EHV-1. Therefore, the new and improved allelic discrimination real-time PCR assay is a very valuable tool for investigating outbreaks of EHV-1 infection. 52
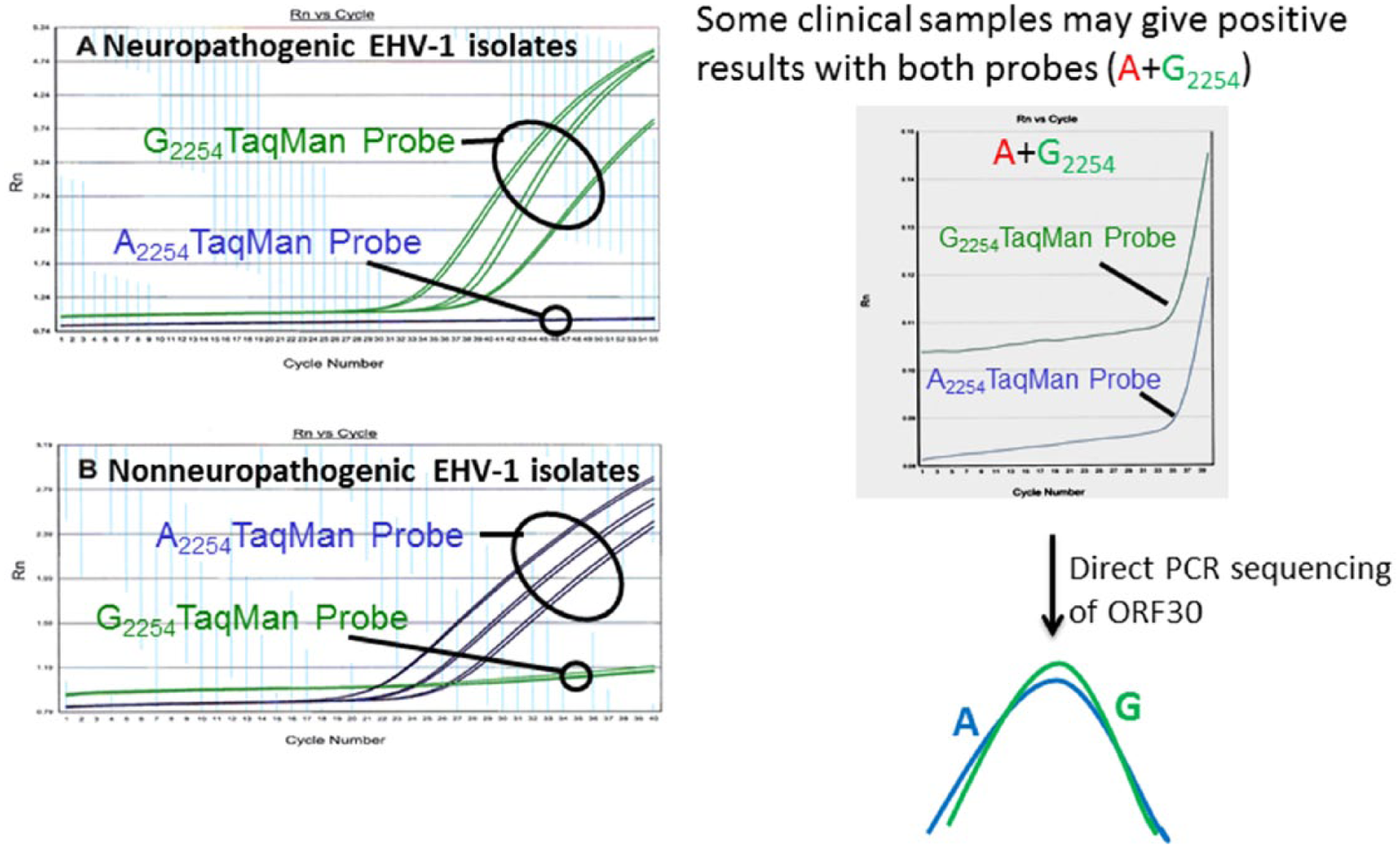
Allelic discrimination real-time polymerase chain reaction (PCR) targeting the open reading frame 30 (ORF30) that encodes viral DNA polymerase enzyme (left panel). Some clinical samples may contain both A2254 and G2254 and cross-react with both probes (right panel). Presence of both genotypes in the clinical samples can be further verified by direct sequencing of the PCR product.
Key questions about detecting A→G2254 substitution (A/G2254 genotype) in clinical specimens
Some studies support an association between EHM and the G2254 genotype described previously. 40 However, there is an increasing body of compelling evidence to indicate that this nucleotide substitution is not the only determinant of enhanced neuropathogenicity. 12 In a previous survey, 41 24% of isolates from horses with neurologic disease possessed the A2254 and not the G2254 genotype. Similarly, we identified a number of A2254 genotype EHV-1 isolates from cases of neurologic disease, as well as the isolates of the G2254 genotype from numerous horses with no evidence of neurological involvement. In addition, we have identified viruses with non-synonymous nucleotide substitutions in ORF30 besides A→G2254, from horses without signs of neurologic disease; this presents the possibility that these may have an attenuating effect on the viral phenotype. Thus, identifying and genotyping EHV-1 field strains using allelic discrimination real-time PCR raise several questions that need to be considered but for which we do not yet have answers (Table 2).
Key questions and answers in relation to detecting A→G2254 substitution (A/G2254 genotype) by allelic discrimination real-time polymerase chain reaction (PCR).*

EHM = equine herpesviral myeloencephalopathy; EHV-1 = Equid herpesvirus 1; ORF = open reading frame; gB, gD = glycoprotein B and D, respectively; LAT = latency-associated transcripts.
Differences in sensitivity of real-time PCR assay and virus isolation and interpretation of results
There is some confusion about interpretation of real-time PCR and VI results in the laboratory detection of EHV-1 infection. The sensitivities of these assays are significantly different, and there is only a single study that compares VI with qPCR using clinical specimens from experimentally inoculated horses (Fig. 2). 41 That study demonstrated that VI could only detect infectious virus ≤5 dpi, whereas qPCR could detect EHV-1 DNA ≤21 dpi in the same nasal swab samples. However, the number of viral DNA copy numbers that could be detected by qPCR decreased significantly from 105–107 per mL at 1–6 dpi to 100–101 per mL at 14–21 dpi. Furthermore, the number of positive horses that could be detected by these assays varied significantly during the course of the infection. The number of horses positive by VI dropped from 87% (1–2 dpi) to 20% by 5 dpi, and virus could not be detected after 6 dpi. In contrast, the qPCR could detect viral DNA in 87% of horses ≤12 dpi following which the percentage rapidly dropped to 53% by 14 dpi and 13% by 21 dpi. The number of horses tested positive by qPCR dropped rapidly starting at 12 dpi. Taken together, these data clearly showed that the number of horses sampled during an outbreak and the type of laboratory test performed can have a significant impact on confirming EHV-1 infection and management of an outbreak. Finally, the interpretation of real-time PCR and VI testing should be done with caution (Table 3). The following points play an important role in interpretation of the respective laboratory test outcomes:
Real-time PCR detects a very small number of viral DNA molecules in clinical specimens, whereas VI requires a higher number of viable virus particles (replication competent) to produce plaques in cell culture.
Real-time PCR detects both infectious virus and noninfectious viral DNA in clinical samples, and therefore has a higher sensitivity compared with traditional VI.
A real-time PCR–positive result does not necessarily mean there is infectious virus in a clinical sample, as only VI can detect infectious virus.
A real-time PCR assay should be the first choice for rapid detection of EHV-1 nasal shedding during outbreaks and for identifying horses and facilities that need to be placed under quarantine.
In addition to PCR testing, it is important to perform VI during outbreaks and archive EHV-1 strain(s) for retrospective molecular characterization and molecular epidemiological studies.
It is not recommended to screen clinically normal horses for the presence of EHV-1 DNA by qPCR or real-time PCR.
Specific studies have not been conducted to determine if the modified live virus (MLV) vaccine strain of EHV-1 is detectable in nasopharyngeal or nasal swabs following vaccination. However, there are reports that the MLV vaccine strain has been identified in aborted fetuses. 51
Suggested actions to be taken based on real-time polymerase chain reaction (PCR) test results.
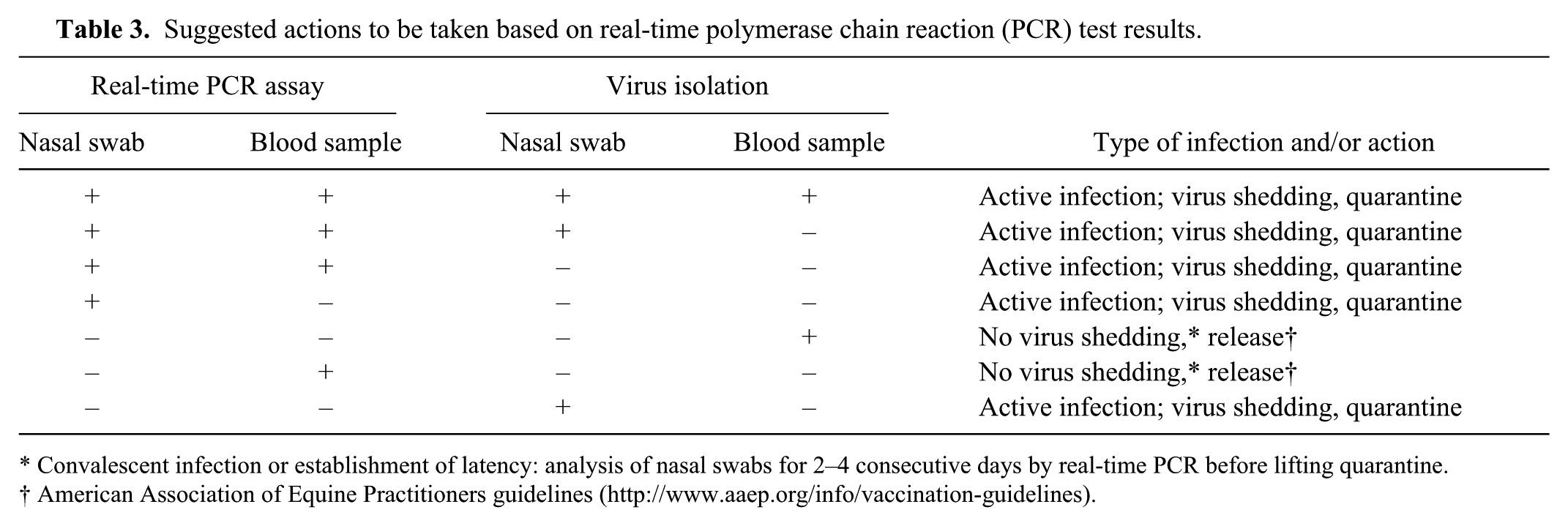
Convalescent infection or establishment of latency: analysis of nasal swabs for 2–4 consecutive days by real-time PCR before lifting quarantine.
American Association of Equine Practitioners guidelines (http://www.aaep.org/info/vaccination-guidelines).
The results of VI and real-time PCR testing of the same sample(s) may not agree with each other. In such situations, the respective findings should be evaluated carefully (Table 3). In addition to this, there are some caveats when using real-time PCR to confirm a clinical diagnosis of EHV-1 infection during disease outbreaks:
All real-time PCR or qPCR assays need to be carefully developed and validated. Real-time qPCR assays need to include appropriate internal controls to normalize for DNA purification and PCR amplification efficiencies.
Results may vary between laboratories because of the use of various nucleic acid extraction methods, target gene, and specific commercial PCR reagents. This variation can be reduced by sending samples to the same laboratory with proven expertise and experience in testing for EHV-1. This is especially important when paired samples from the same animal (taken at 2 different times) or from the same outbreak are submitted for laboratory testing.
Sensitivity and specificity of real-time PCR assays can be affected by a variety of factors such as sample type, sample volume, viral nucleic acid extraction method, target gene, primers and probes and their concentrations, commercial PCR reagent kits, number of cycles, and cutoff point. Thus, harmonization of real-time PCR protocols between laboratories is very important.
The threshold cycle (Ct) values can be used to indicate the approximate viral DNA concentration in samples: Ct < 25 = high (acute stage of infection); Ct 25–30 = moderate; Ct 30–35 = low; Ct 35–40 = suspect. However, this classification varies among laboratories because such ranges are based on specific assay conditions (e.g., reagents, primers, probes, target genes) and validation.
The real-time qPCR should be used for better characterization of the stage of the disease, assessment of risk of exposure to other horses, monitoring of response to treatment, and in research studies.
In summary, direct methods of detection are preferred over indirect methods. Of the direct methods, real-time PCR–based assays are more sensitive than the traditional VI method, which is predicated on the presence of live virus in clinical specimens. As indicated above, these 2 methods have respective advantages and disadvantages (Table 4). The main disadvantage of molecular diagnostic assays such as real-time PCR is the unavailability of the virus for further studies. Presently, VI from clinical samples is attempted by a decreasing number of laboratories. Positive PCR results does not necessarily equate with the risk of naturally infected horse transmitting infection to a susceptible cohort.
Specificity and sensitivity of various assays and their applicability in routine laboratory testing for the detection of Equid herpesvirus 1 (EHV-1).*
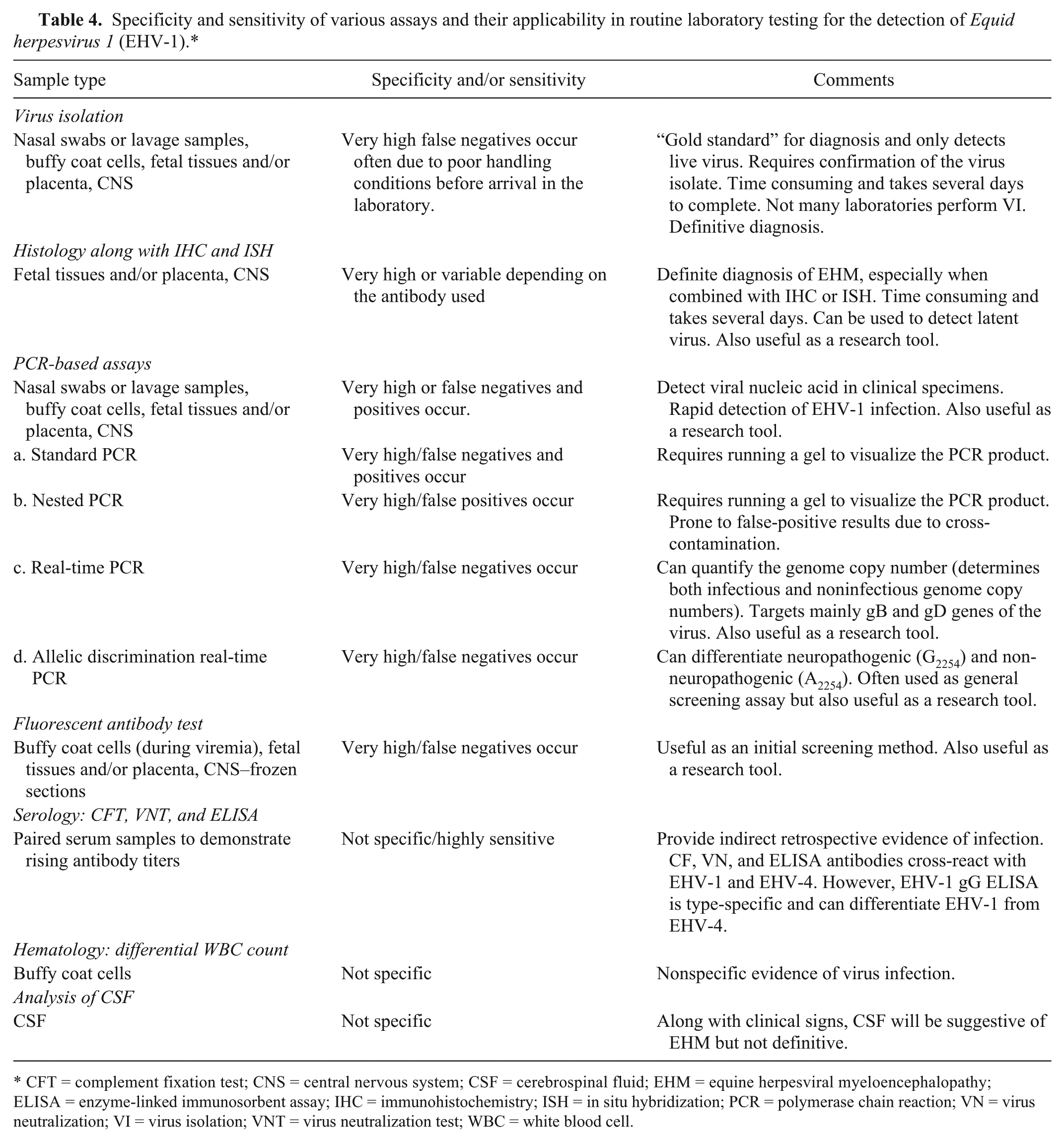
CFT = complement fixation test; CNS = central nervous system; CSF = cerebrospinal fluid; EHM = equine herpesviral myeloencephalopathy; ELISA = enzyme-linked immunosorbent assay; IHC = immunohistochemistry; ISH = in situ hybridization; PCR = polymerase chain reaction; VN = virus neutralization; VI = virus isolation; VNT = virus neutralization test; WBC = white blood cell.
6. Insulated isothermal PCR assay
A hydrolysis probe–based insulated isothermal PCR (iiPCR) assay for the detection of EHV-1 and other equine viral pathogens has been developed; this assay has a very high specificity and sensitivity compared to real-time PCR. Fluorogenic probe hydrolysis chemistry is used to generate a fluorescent signal when a specific DNA sequence of EHV-1 is amplified. This assay is specially designed to be used on a compatible iiPCR instrument.10,56 The instrument is intended for use by trained personnel in veterinary clinics, racetracks, breeding facilities, and diagnostic laboratories.
7. Loop-mediated isothermal amplification assay
Loop-mediated isothermal amplification (LAMP) assays for the detection of EHV-1 nucleic acid in equine nasal swab samples have been described. 39 The LAMP assay can be carried out under isothermal conditions (60–65°C), and the amplification of nucleic acid can be judged by the naked eye based on the turbidity or fluorescence of the reaction mixture. The rapidity and simplicity of this assay allow it to be used in clinical laboratories, although these assays need to be validated properly. A new version of the LAMP assay has been described without using DNA extraction from nasal swab samples. 39 The omission of the DNA extraction step saves cost, time, and the labor needed for nucleic acid extraction.
8. Pyrosequencing
A pyrosequencing technique has been used to detect and distinguish neuropathogenic and non-neuropathogenic strains of EHV-1 in paraffin-embedded tissues from cases of equine abortion and EHM. 54 Pyrosequencing allows sequencing of the PCR amplicon and immediate genotypic (A2254 or G2254) characterization of the virus. To date, the technology has not been evaluated for routine testing of EHV-1 suspect samples.
9. In situ hybridization assay
In situ hybridization (ISH) is a powerful technique that allows for localization of nucleic acid targets in fixed tissues and cells. The principle of ISH is that nucleic acids, when preserved adequately in a histologic specimen and when fixed appropriately, can be detected by using a nucleic acid probe. ISH has been used to detect both active infection, as well as latent EHV-1 infection, in tissues.6,11,24,38,48,50,53 Radiolabeled or digoxigenin-labeled DNA and RNA probes have been used to detect viral DNA or messenger RNA transcripts (e.g., latency associated transcripts), respectively, in tissue sections. However, ISH assays cannot differentiate between the current commercial MLV vaccine and natural occurring EHV-1 strains circulating in the field.
III. Indirect demonstration of EHV-1 infection
A. Serology—detection of antibodies
Demonstration of a humoral antibody response to EHV-1 by serological assay is one method to demonstrate exposure to EHV-1. At least 4 serological assays have been described for the detection of antibodies to EHV-1: complement fixation test (CFT), virus neutralization test (VNT), agar gel immunodiffusion (AGID) assay, and ELISA. 59 However, because of preexisting antibodies to either EHV-1 or EHV-4 or to both viruses as the result of prior infection or vaccination, serologic assays are less useful as a diagnostic tool. Serologic detection of EHV-1 infection can be achieved by demonstration of seroconversion or a 4-fold rise in antibody titer between paired sera taken during acute and convalescent stages of the disease (sera collected at onset and 2–4 weeks later; see below). Serum from mares that abort or from horses with EHV-1 neurologic disease may already contain peak levels of antibodies, and no increase in titers may be detectable in sera collected subsequently. Serum antibody levels to EHV-1 can be determined by CFT, VNT, or ELISA. The presence of CF and VN antibodies in equine serum does not differentiate EHV-1 from EHV-4 infection. The demonstration of seroconversion or a 4-fold or greater rise in antibody titers to EHV-1 between paired sera by any of these tests is serologic confirmation of recent infection with the virus. Herd-based testing for EHV-1 or EHV-4 “outbreaks” may be possible by testing clinically affected horses and comparing titer levels to cohorts in the same herd without evidence of respiratory disease. However, there are no internationally recognized reagents or standardized laboratory protocols for performing these tests and, as a result, there is the potential for frequent variation in serologic results among laboratories.
1. Complement fixation test
The complement fixation test is usually not used for the routine detection of EHV-1 because it is fairly difficult to perform, requiring qualified, trained personnel to achieve reliable and consistent results. The test is based on the principle that complement is fixed during an antigen–antibody reaction. Complement-fixing antibodies generally appear early and persist for short periods, being of limited value for seroepidemiological studies. However, they are very specific in primary infections, enabling the determination of the infecting EHV type (e.g., EHV-1 or EHV-4), as demonstrated by the monotypic responses observed in primary infections. The CFT generally measures the immunoglobulin M (IgM) response to EHV-1. The IgM antibodies appear at 4–5 dpi and peak ~20–30 dpi, decreasing to baseline values usually within 60–80 dpi. A high CF antibody titer in a single serum sample is suggestive of an acute or recent infection and is a valuable initial diagnostic test in suspected respiratory or EHM cases. Similarly, seroconversion or a 4-fold rising CF antibody titer between paired serum samples taken 14–28 days apart provide unequivocal evidence of EHV-1 infection.
2. Virus neutralization test
Virus neutralization test (or serum neutralization) is labor intensive and requires highly qualified and trained personnel to achieve consistent results. Horses mount a strong IgG immune response to EHV-1, detectable at 8–10 dpi, peaking at 30–40 dpi and persisting for many months (>9 months). VNT principally measures the IgG immune response to EHV-1. The IgG response to EHV-1 is highly specific and only neutralizes EHV-1. In contrast, the IgG response to EHV-4 neutralizes both EHV-1 and EHV-4 (cross-reactive antibodies generated to epitopes common both to EHV-1 and EHV-4). The longevity of VN antibodies lessens their usefulness for investigation of acute EHV-1 infections. Because VN antibodies are long lived after initial infection, they have a valuable application in serologic surveys. Demonstration of seroconversion or a 4-fold rise in VN antibody titers between paired serum samples taken 14–28 days apart provides unequivocal evidence of EHV-1 infection.
3. Enzyme-linked immunosorbent assay based on glycoprotein G
ELISA is simple, easy to perform, and can be used to screen large numbers of serum samples for EHV-1 antibodies. However like the VNT, most ELISAs detect IgG response and hence cross-react with antibodies to both EHV-1 and EHV-4 (not type-specific). To overcome this issue, a type-specific ELISA targeting glycoprotein G (gG) was developed.16,21 This ELISA, targeting at least 1 type-specific epitope in gG, can discriminate between the IgG immune response to EHV-1 and EHV-4. 13 Both type-common and type-specific ELISAs are commercially available. The type-specific commercial ELISA a based on gG is more useful for distinguishing EHV-1 and EHV-4 infections in horses, and could be used to demonstrate a 4-fold rise in antibody titer between paired serum samples taken 14–28 days apart (i.e., provide unequivocal evidence of EHV-1 infection).
4. Other ELISAs (experimental peptide ELISAs)
A unique immunodominant region in the gE of EHV-1 has been identified (amino acids [aa] 169–201), and antibodies to EHV-4 failed to react with this region in ELISAs. 5 Similarly, a unique immunodominant region in the gG of EHV-4 has been identified that does not cross-react with antibodies to EHV-1. An ELISA using 2 synthetic peptides, EHV-1 gE (aa 169–188) and EHV-4 gG (aa 319–330), previously identified as the major EHV-4–specific epitope in gG, has been used specifically to detect antibodies to EHV-1 and EHV-4, respectively. 5
Furthermore, a peptide pair derived from EHV-1 gE and EHV-4 gG has been evaluated using acute and convalescent sera from horses infected experimentally and naturally as well as a panel of field horse sera. 34 The peptide ELISA was able to identify horses that had been infected with EHV-1 or EHV-4 based on the results using acute and convalescent sera collected from natural outbreaks. The assay proved robust with respect to determining EHV-1 and EHV-4 antibody status. Also, the peptide ELISA was able to detect type-specific seroconversion for EHV-1 in vaccinated animals. With further validation, the EHV-1/EHV-4 peptide ELISA described in this study could serve as a reliable and cost-effective alternative to current methods for serological EHV-1 and EHV-4 diagnosis. 34 However, both of these peptide ELISAs must be further validated with a large number of equine serum samples before being accepted for use in routine testing.
5. Agar gel immunodiffusion assay
An AGID assay using a baculovirus expressed recombinant EHV-1 gG protein has been reported. 21 Compared with VNT, the AGID assay showed very good specificity (100%) and sensitivity (99.5%). However, this assay could not discriminate between antibodies against EHV-1 and EHV-4.
Major points to consider when using serologic assays
Serologic assays indicate indirect evidence of EHV-1 infection.
The CFT does not differentiate between IgM and IgG, and may measure both.
Most of these serologic tests detect antibodies that are cross-reactive between EHV-1 and EHV-4.
Only the gG-specific ELISA can distinguish EHV-1 from EHV-4.
Paired serum samples (14–28 days apart) must be tested to demonstrate seroconversion or rising antibody titers.
Maternal antibodies and antibodies stimulated by vaccine strains of EHV-1 cannot be distinguished from antibodies from natural infection.
B. Hematology—analysis of white blood cell counts
EHV-1 and EHV-4 can cause biphasic changes in total and differential white blood cell (WBC) counts. There is an initial transient leukopenia and lymphopenia at 7–9 dpi. This is followed by leukocytosis and lymphocytosis ≤21 dpi. However, there is significant variation in normal WBC counts among horses, and other viral diseases (e.g., equine viral arteritis) can cause similar hematological changes. In summary, changes in WBC counts cannot be used as evidence of EHV-1 infection.
C. Cerebrospinal fluid analysis
Cerebrospinal fluid of horses with EHM may have increased protein (mainly albumin) concentration. CSF also exhibits yellow discoloration (xanthochromia) because of increased breakdown of red blood cells and increased protein. The presence of characteristic neurologic signs in combination with CSF changes is indicative but not confirmatory of EHM. Antibodies to EHV-1 can be detected in the CSF as a result of leakage from the blood vessels where endothelial cells are damaged (infection and vasculitis). Presence of EHV-1 antibodies in CSF is not per se conclusive evidence of EHM. CSF changes should be supported by other laboratory tests (e.g., VI and/or real-time PCR) before identification of EHM is definitive.
IV. Detection of EHV-1 during latent infection
EHV-1 establishes an asymptomatic latent infection in sensory ganglia, lymphoid tissues (particularly those draining the respiratory tract), and cluster of differentiation (CD)5+/CD8+ lymphocytes.4,49 Latency occurs when the entire viral genome is present in the infected cell but only a limited number of genes are transcribed and expressed. Specifically, latency-associated transcripts (LAT) have been detected by PCR and ISH. However, antemortem clinical and laboratory confirmation of latent EHV-1 infection is extremely difficult and challenging for the following reasons.
Latently infected horses show no clinical signs of EHV-1 infection or disease.
EHV-1 establishes latent infection in a limited number of cells as indicated above.
Latently infected cells will not express major viral proteins (antigens) and, therefore, cannot be detected by any of the antigen-detection assays described above.
Latently infected cells contain a relatively low copy number of the viral genome.
During latent infection, with the exception of the LAT genes, most of the viral genes are not expressed.
Thus, EHV-1 cannot be detected in latently infected horses (antemortem) by most of the previously described laboratory tests (Table 5). However, all of these assays can detect virus following reactivation of the latent infection (results will be very similar to an active lytic infection). Presumptive detection of latent EHV-1 infection in a horse can be made by:
Demonstration of virus in buffy coat cells by co-cultivation. This is the gold standard because it unequivocally demonstrates the presence of reactivatable latent infection. However, a negative result by co-cultivation does not indicate the absence of latent infection.
Demonstration of expression of LAT genes in cells by PCR. However, this is technically challenging and these genes are not universally expressed in all cells carrying latent virus.
Demonstration of PCR-positive buffy coat cells without any clinical signs, and virus shedding in nasal swabs as determined by VI and PCR.
Detection of viral DNA by PCR is not unequivocal evidence of latent infection (reactivation of latent infection can give similar results).
Use of qPCR to demonstrate virus copy number (comparative analysis). This was proposed as an alternative approach to the detection of LAT in latently infected cells, but this method will not provide definitive detection of latent infection.
Detection of VN antibody titers without any clinical signs.
Detection in formalin-fixed tissue sections using RNA–RNA ISH, which detects the expression of LAT genes, and RNA–DNA ISH, which detects the viral genome. Demonstration of latent infection in autopsy samples is more straightforward than in antemortem samples.
Commonly used laboratory tests for the detection of lytic and latent Equid herpesvirus 1 (EHV-1) infection.

White blood cells.
Cocultivation with susceptible cell for extended period of time (may require up to 3 passages). The sensitivity of the co-cultivation assay can be increased by treatment of buffy coat cells with T-cell mitogens (phytohemagglutinin [PHA] and pokeweed mitogen [PWM]), or by using immune suppressant (dexamethasone) in tissue culture medium.
Brain and spinal cord with microscopic lesions.
May be transiently positive during viremia.
Latent infection can be maintained longer than the duration of serum-neutralizing antibodies in serum. During reactivation and lytic infection, complement fixation and serum neutralization antibody titers increase.
Footnotes
Acknowledgements
We thank Ms. Kathleen M. Shuck for critical reading of the manuscript.
Authors’ contributions
UBR Balasuriya contributed to conception and design of the study, and drafted the manuscript. All authors contributed to acquisition, analysis, and interpretation of data; critically revised the manuscript; gave final approval; and agree to be accountable for all aspects of the work in ensuring that questions relating to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
a.
SVANOVIR EHV1/4 ELISA, Svanovir Biotech, Uppsala, Sweden.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by funds from the Kentucky Agricultural Experiment Station Hatch Project (KY041042), Grayson Jockey Club Research Foundation, and contracts awarded to Dr. Udeni B. R. Balasuriya at the Maxwell H. Gluck Equine Research Center, Department of Veterinary Science, College of Agriculture, Food and Environment, University of Kentucky.