
Abstract
Mannheimia haemolytica is a major bacterial component of bovine respiratory disease (BRD); unfortunately, very little is known about M. haemolytica transmission dynamics among cattle. Identifying potential variation in M. haemolytica populations over time and induction of nasopharyngeal colonization and subsequent shedding are 2 areas where knowledge is lacking. In our study, 2 separate loads of 20 mixed-origin, male calves were purchased through an order buyer on different dates. Deep nasopharyngeal cultures (NPC) were performed on all calves on arrival and, if M. haemolytica–negative, a second screening culture was obtained. Calves that were negative on 2 initial NPCs (NEG; n = 4) were subsequently challenged with a previously isolated field strain of M. haemolytica in both the upper and lower respiratory tract, individually housed, and then monitored for M. haemolytica shedding via NPCs at 0.5, 1, 3, 5, 7, and 9 days postchallenge. Naturally M. haemolytica–positive calves (2 per load) were kept for additional daily cultures (POS; n = 4). Individual calf M. haemolytica status for both the POS and NEG groups was inconsistent between study days. Additionally, pulsed-field gel electrophoresis performed on isolates from the positive cultures showed that the NEG calves did not shed the M. haemolytica challenge strain, but rather 2 distinct clusters of M. haemolytica were shared among POS and NEG calves regardless of their initial status. Although sample sizes were small, these findings illustrate how variable the results of a single nasopharyngeal swab can be and the challenges of using an individual culture to truly represent animal M. haemolytica status.
Introduction
Bovine respiratory disease (BRD) is widely considered the most common and costly postweaning beef cattle disease.17,18,25 Although BRD is truly a multifactorial disease with bacterial, viral, environmental, and host factors involved, Mannheimia haemolytica is the most frequently isolated bacterial pathogen from BRD cases.16,17,24 Mannheimia haemolytica has many virulence factors that have been identified, and M. haemolytica may transition from being a commensal organism within the nasal passages into a pulmonary pathogen. 16 Unfortunately, it is unclear how M. haemolytica makes this transition, and whether a specific M. haemolytica isolate is likely to transfer from calf to calf within a population. A variety of methods have been used to explore M. haemolytica population dynamics over the years including serotyping, 1 plasmid typing, 7 and selective culture 15 with pulsed-field gel electrophoresis (PFGE) gaining popularity because of its high discriminatory power and medium-to-high repeatability.18,24
Because definitive antemortem diagnosis of BRD based on clinical signs can be difficult, 4 additional diagnostic aids such as nasal cultures, transtracheal cultures, transtracheal washes, and bronchoalveolar lavages have been used in both research and clinical settings.2,6,9,10 Although several studies13,15,19 have attempted to use nasopharyngeal culture (NPC) as a way to describe the population of M. haemolytica within individual calves and how it moves within a group of calves, it remains an imperfect test. Other studies have attempted to correlate NPC results with cultures obtained elsewhere in the respiratory tract with mixed results.2,8,10,15,24 Although inoculation of calves with M. haemolytica can induce BRD,7,14 a repeatable model for M. haemolytica inoculation that results in subsequent shedding has not been established.
The objectives of our pilot study were to describe the arrival M. haemolytica status in a group of calves via NPC, identify negative animals (NEG), and evaluate the ability of an artificially inoculated M. haemolytica strain to colonize and shed from those negative calves. Additionally, this pilot study sought to identify healthy, naturally M. haemolytica–positive calves (POS) on arrival and evaluate the shedding patterns of M. haemolytica in those calves via NPC.
Materials and methods
Calf selection and management
Two loads of 20 male calves were procured through an order buyer from local livestock auction markets and transported to the Kansas State University Large Animal Research Center (Manhattan, Kansas; total n = 40). Within a few hours of arrival, all calves were weighed and individually identified with ear tags. Additionally, the nasopharynx of each calf was cultured by passing a single guarded calcium alginate swab a through the right external nares. The guarded swab was then guided into the caudal nasal passage where the inner swab was passed through the guard, rotated against the nasal mucosa, and retracted back into the guard prior to removal from the nasal passages. The swab was placed immediately into sterile transport media b and either temporarily refrigerated or submitted directly to the Kansas State Veterinary Diagnostic Laboratory (Manhattan, KS) for aerobic culture. None of the calves received antimicrobial treatment for the control of BRD, vaccinations, or implants at any time during the study, and any animal that required antimicrobial treatment for clinical disease was removed from the study. Based on the results of the initial screening culture(s), calves were segregated into culture-positive (POS) and culture-negative (NEG) groups (Fig. 1).
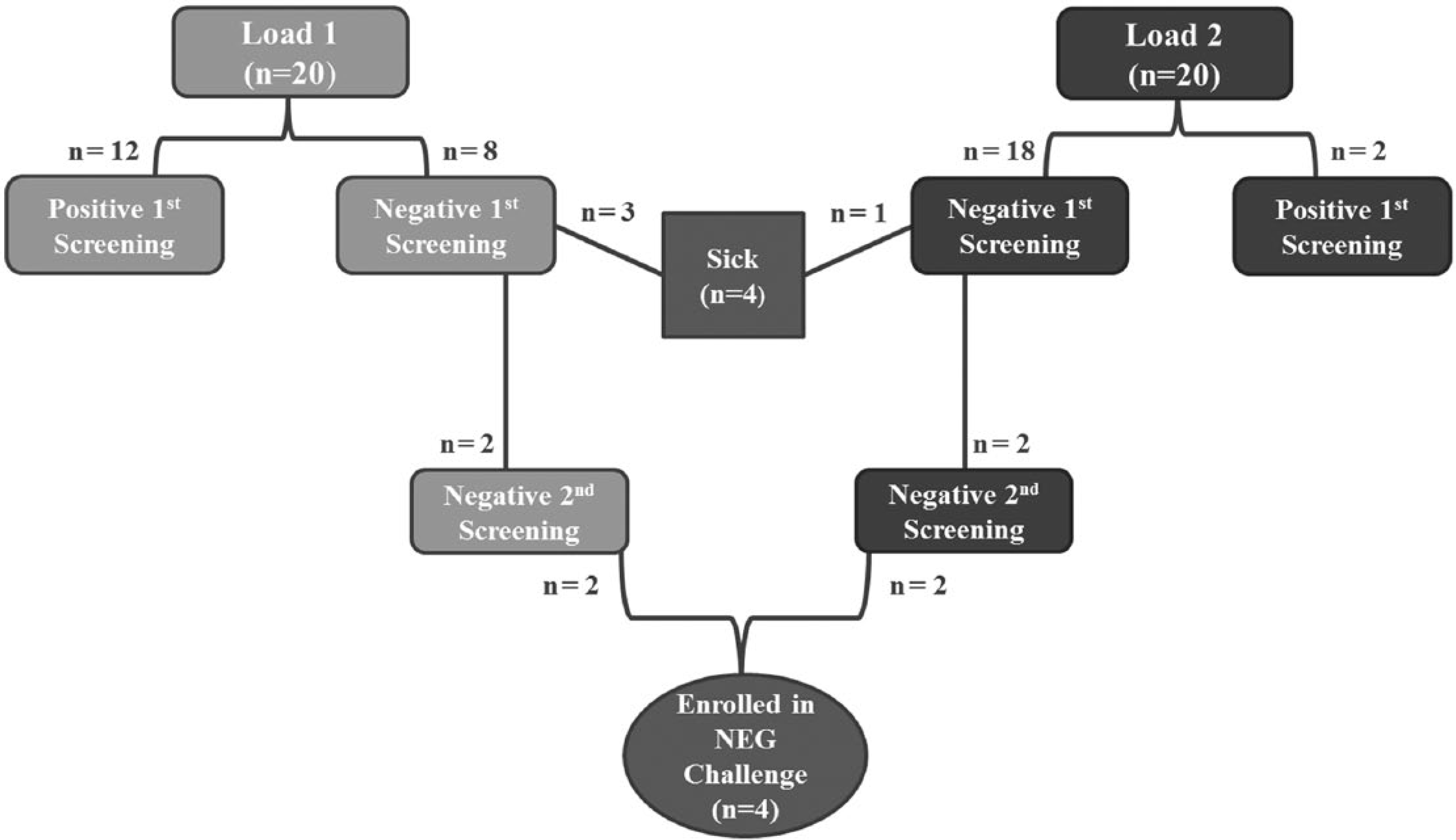
Summary of Mannheimia haemolytica screening cultures performed on each load of calves and selection of calves for inclusion within the final NEG challenge pilot. NEG = negative for M. haemolytica on 2 screening nasopharyngeal cultures interpreted in parallel.
During the prechallenge period, all calves were housed in open-air, dirt-floor group housing pens with a total area of 297 m2 per pen and provided with an adequate amount of bunk space, access to water, and shelter. In order to prevent any possible nose-to-nose contact, calves were individually housed immediately following challenge in either identical open-air, dirt-floor pens or in identical 13.4-m2 stalls. Within the 2 types of housing, the challenged calves were managed similarly. Throughout the trial, calves were fed a diet consisting of a grain mix with grass hay and water made available ad libitum. Calf management and sampling procedures described were part of a protocol approved by the Kansas State University Institutional Animal Care and Use Committee.
Preparation of challenge media
A M. haemolytica isolate obtained from a field case of BRD and used in a prior challenge study 5 was grown on a 5% ovine blood agar plate in a CO2 incubator at 37°C for 18–22 hr. Isolated colonies were inoculated into brain–heart infusion broth and incubated for 16–18 hr at 37°C on a rotary shaker. The bacteria were then pelleted via centrifugation at 15,000 × g for 20 min, washed 3 times in sterile phosphate buffered saline (PBS), and re-suspended in sterile PBS to an optical density (OD) of 1.8–2.0 at 600 nm. This OD corresponds to a bacterial concentration of 3 × 108 colony forming units (CFU)/mL as determined by a standard curve of M. haemolytica CFUs versus OD at 600 nm.
Challenge and sampling of negative calves
Four NEG calves were restrained in a chute with their heads elevated and stabilized using a rope halter. A 5.9-mm endoscope with 2-mm biopsy channel c was introduced into the right nasal passage of each calf and passed through the nasopharyngeal region and laryngeal folds into the trachea and advanced to the right cranial tracheobronchus. A 140-cm polyurethane catheter was then passed through the endoscopic biopsy channel, and 10 mL of M. haemolytica inoculum (3 × 108 CFU/mL in PBS) followed by 60 mL of sterile PBS was administered. Following the endoscopic challenge, calves were also inoculated with 2 mL of M. haemolytica (3 × 108 CFU/mL in PBS) deposited into the right nasal passages via a syringe.
Using the deep nasopharyngeal sample collection technique described above, samples for culture were collected at 0.5, 1, 3, 5, 7, and 9 days postchallenge from the right nasopharyngeal area. Rectal temperatures were also recorded at the time of each NPC. Potential shedding of the challenge organism was determined by comparing the PFGE pattern of a single M. haemolytica isolate from each positive culture plate with the known challenge strain.
Challenged calves were euthanized via a penetrating captive bolt d according to the American Veterinary Medical Association Guidelines (https://www.avma.org/KB/Policies/Documents/euthanasia.pdf), and a full autopsy was performed 10 days postchallenge. During autopsy, the lungs were removed intact and then weighed; lung lesions were scored by an experienced veterinarian using a standardized system 12 similar to previous work.4,5,23
Monitoring of naturally M. haemolytica–positive calves
Four naturally POS calves (2 from each load) were retained after initial screening for further monitoring of NPC status. Prior to the start of daily cultures, all 4 calves were confirmed culture positive for M. haemolytica, and up to 10 isolates were selected and frozen per calf for later PFGE comparison. If <10 distinct colonies were present on the plate, all M. haemolytica isolates were selected from the plate and frozen for later PFGE comparison. Once the second confirmatory culture results were obtained, NPCs were performed daily for 3 days as the calves continued to be monitored for clinical signs of illness. Single isolates were retained from each positive daily culture and examined by PFGE.
Clinical illness scores
All calves were observed by the same veterinarian (SF Capik) twice daily throughout the study (morning and evening) for any signs of illness and assigned a clinical illness score (CIS) based on the following criteria: 0 = normal calf, 1 = mild signs of depression, 2 = moderate depression, 3 = severe depression, and 4 = severe prostration and/or recumbence. 21 Following arrival and prior to challenge, any calf with a CIS of >0 was evaluated by a veterinarian and appropriate treatment applied. If antimicrobial treatment was deemed necessary at any time after arrival, the animal was treated and excluded from the study. Any calf given a clinical illness score of 4 at any point during the trial was humanely euthanized and an autopsy performed.
Microbiology culture methods
Nasopharyngeal swabs were rolled directly onto trypticase soy agar plus 5% sheep blood (SBA) and chocolate agar plates. Using a sterile loop, multiple passes (in a single continuous streak) were made through the inoculated area of the agar plates. A second sterile loop was used, in the same manner, from the second to third quadrants of the agar plate so that isolated colonies would be present. The SBA and chocolate plates were incubated at 37°C in 5% CO2 for 18–24 hr. Following incubation, plates were examined, and colonies suspected to be M. haemolytica were either re-streaked for isolation or identified directly (depending on degree of colony isolation and phase of project). For the second confirmatory culture of POS calves in which up to 10 isolates were selected for PFGE, isolates were chosen preferentially from colonies that were either 1) phenotypically different or 2) distinct from background growth. All bacterial identification was performed using matrix-assisted laser desorption ionization e according to the manufacturer’s instructions. Isolates, in pure culture, were stored in Brucella broth plus 10% glycerol at −80ºC for further PFGE analysis.
Pulsed-field gel electrophoresis
Actively growing, pure bacterial cultures on blood agar plates were collected using sterile cotton swabs and resuspended in a buffered resuspension solution (100 mM Tris–HCl, pH 8.0, and 100 mM ethylenediamine tetra-acetic acid [EDTA]). Cell concentration was adjusted to 0.72 units, f and 200 μL of the cell suspension was mixed with 200 μL of 1.2% agarose in water and proteinase K at 0.5 mg/mL g and dispensed into plug molds. h Bacteria-containing agarose plugs were lysed in the presence of 50 mM Tris–HCl (pH 8.0), 50 mM EDTA, 1% sarkosyl, and 0.32 of mg/mL proteinase K at 54°C with constant agitation. Plugs were successively washed with distilled water and 10 mM Tris–HCl (pH 8.0) and 1 mM EDTA at 50°C with constant agitation. Agarose slices were digested with SmaI restriction enzyme i for 2 hr according to manufacturer’s instructions, and electrophoresis was performed j on a 1% agarose gel k with 0.5× Tris–borate–EDTA buffer (50 mM Tris–HCl, pH 8.4, 45 mM boric acid, and 0.5 mM EDTA) at 14°C. Salmonella enterica subsp. enterica serovar Braenderup ATCC BAA-664 was used as a molecular weight marker strain, and plug DNA from that organism was digested with XbaI. l The molecular weight marker DNA was interspersed in lanes at regular intervals across each agarose gel in order to normalize banding patterns both within and between agarose gels. Electrophoresis conditions were Block 1: Initial Switch Time 2.0 sec, Final Switch Time 5.0 sec, Run Time 12 hr; Block 2: Initial Switch Time 2.0 sec, Final Switch Time 5.0 sec, Run Time 12 hr; for both blocks: colts/cm: 5.6, V, Included Angle: 120°, 24 hr total. Agarose gels were stained with 1 µg/μL of ethidium bromide, destained, and an image obtained. m Clustering analysis was performed n using the Dice coefficient and the unweighted pair group method with arithmetic mean clustering method with settings for optimization: 1.0%, band tolerance: 1.5%. A 90% similarity threshold cutoff value was used when comparing the challenge strain and the cultured isolates. 26
Results
The average weights of load 1 (arrived on 06/10/2013) and load 2 (arrived on 06/14/2013) were 169.3 kg and 165 kg, respectively. Between the 2 loads, a total of 32 bulls and 8 steers were enrolled in the study (Fig. 1). After the initial screening culture, only 26 culture-negative calves remained eligible for the second screening culture. Following the second screening culture, 4 calves remained culture-negative (2 from each load) and were retained for challenge (NEG). Additionally, 4 culture-positive calves (2 from each load) were kept for additional monitoring (POS).
NEG calves
Following an observation period of 7–10 days in which the NEG calves (n = 4) were determined to be healthy, challenge was performed on trial day 0. Throughout the 10-day postchallenge period, only 1 calf (no. 75) received a clinical illness score of 1 on trial days 3, 4, 7, 8, and 9 after showing signs of mild depression and heat stress. All other calves appeared clinically normal throughout the trial, and no signs of respiratory disease were noted. Although rectal temperatures did not directly correlate with CIS or culture results, it was noted that calf 75 had a consistently elevated rectal temperature (>40°C) throughout the trial. Two other calves (23 and 54) had elevated rectal temperatures (>40°C) on trial day 0.5, and calf 23 also had an elevated rectal temperature (>40°C) on trial day 3.
Postchallenge NPCs revealed consistent results for 3 of the 4 negative calves but inconsistent shedding and day-to-day variability in 1 calf (Table 1). The use of PFGE analysis on a single isolate taken from each M. haemolytica–positive culture plate showed 2 distinct clusters of M. haemolytica (cluster A and cluster B) using a 90% similarity threshold cutoff value. 26 Comparison of cluster A and cluster B to the challenge strain using the same 90% similarity threshold cutoff value revealed that the challenge strain was unrelated to all M. haemolytica isolates selected from study animals (Fig. 2). Additionally, within the 3 calves that cultured positive after challenge, calves 54 and 23 shed isolates that fell within cluster B while calf 75 shed isolates that were consistent with cluster A.
Nasopharyngeal culture results over time from NEG calves (n = 4) challenged with Mannheimia haemolytica via both intranasal and endoscopic routes at time 0.*

NEG = negative for Mannheimia haemolytica on 2 screening nasopharyngeal cultures interpreted in parallel; + = positive; – = negative.
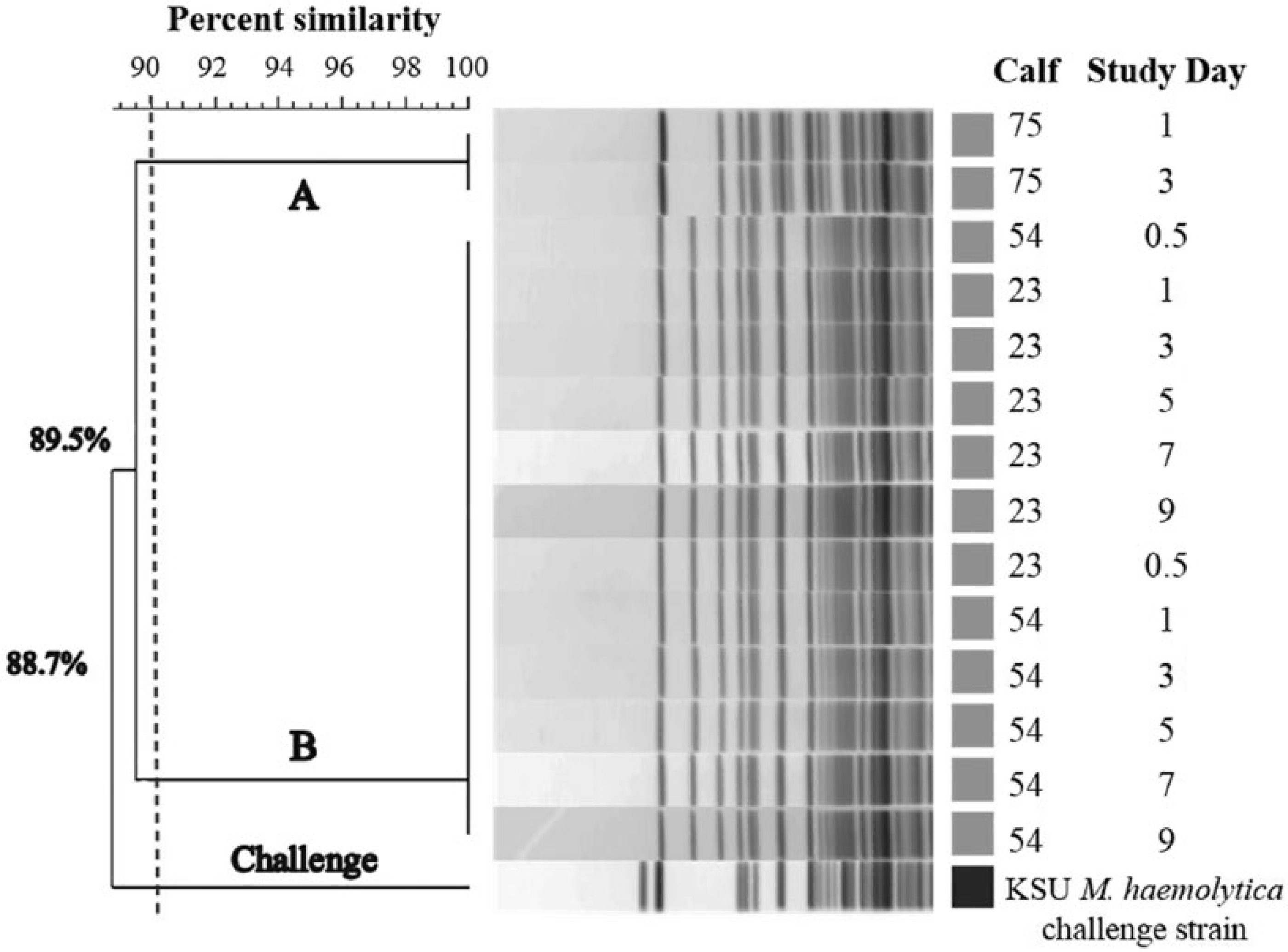
Pulsed-field gel electrophoresis dendrogram and cluster analysis for all Mannheimia haemolytica–positive nasopharyngeal cultures following challenge of NEG calves (n = 4). A single isolate was obtained from each M. haemolytica–positive culture plate and compared to the Kansas State University (KSU) M. haemolytica challenge strain using a 90% similarity threshold cutoff value. Two distinct clusters of M. haemolytica were identified as denoted by the A and B. NEG = negative for M. haemolytica on 2 screening nasopharyngeal cultures interpreted in parallel.
All 4 NEG challenged calves had prominent right cranioventral pulmonary lesions. Among the calves, total consolidation ranged from 10.1% to 37.6% with a median lung score of 19%, and lung weight as a percentage of body weight ranged from 0.93% to 1.47% with a median of 1.04%. Pleural adhesions were present in 3 of the 4 calves and 1 calf (no. 75) had reactive tracheobronchial lymph nodes. Despite gross evidence of BRD consistent with previous M. haemolytica challenge models, bacterial cultures of fresh lung samples from all 4 calves were negative for M. haemolytica.
POS calves
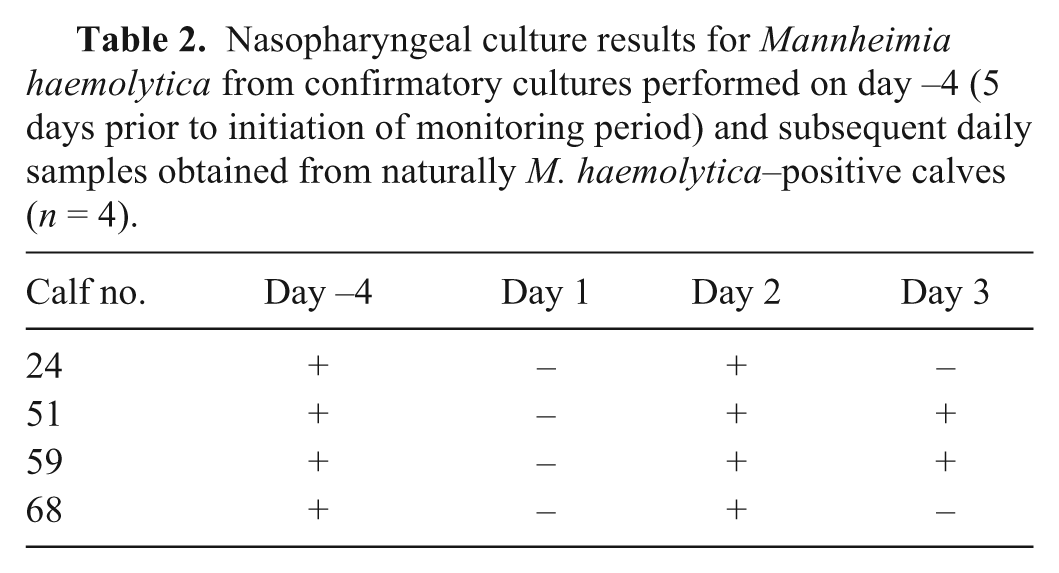
All 4 naturally M. haemolytica–positive calves remained clinically healthy throughout the monitoring period. Despite each of the calves having 2 prior positive cultures, the culture results from the 3 days of monitoring were inconsistent (Table 2). The PFGE analysis of the multiple isolates taken from the second confirmatory culture (calf 24: 10 isolates; calf 51: 1 isolate; calf 59: 10 isolates; calf 68: 5 isolates), and the single isolates taken from the daily culture fell within the same cluster A and cluster B equivalent to those seen in the NEG calves. Additionally, calf 59 had isolates that were consistent with both cluster A (9/10 isolates) and cluster B (1/10 isolates) on the second confirmatory culture. The isolates obtained from calf 51 alternated between cluster A and cluster B depending on the sampling day.
Nasopharyngeal culture results for Mannheimia haemolytica from confirmatory cultures performed on day −4 (5 days prior to initiation of monitoring period) and subsequent daily samples obtained from naturally M. haemolytica–positive calves (n = 4).
Discussion
The fluctuations in culture status and cluster seen within all phases of our pilot study raise questions about both the pattern of M. haemolytica shedding and the ability of an individual culture swab to give a true representation of the M. haemolytica status of a calf. Prior work 20 found M. haemolytica present only on the surface of the nasal epithelium and indicated that a negative nasal swab culture did not necessarily mean that M. haemolytica was absent from the nasal cavity. Other work also showed that M. haemolytica was isolated irregularly even when both right and left sides of the anterior and posterior nasal meatuses of calves were swabbed multiple times a day for several days in a row. 19 The results of our study seem to support these previous studies, as the culture status of each calf within the NEG and POS groups was not always consistent even when daily samples were taken.
Given the multifactorial nature of BRD, the limitations of available diagnostic tests, and the knowledge gaps that exist regarding the hypothesized conversion of M. haemolytica from commensal to pathogenic, the prevalence of M. haemolytica in calves on arrival at a facility can be difficult to establish. Our study found considerable differences in the results of the arrival screening cultures performed on the 2 separate loads of calves we received, with load 1 having an apparent prevalence of 60% and load 2 having an apparent prevalence of 10% (Fig. 1). However, when the second screening culture was performed several days later on previously culture-negative calves, the cumulative apparent prevalence for each load became 88.2% and 89.5%, respectively. The drastic change in apparent prevalence seen in load 2 may reflect the limitations of a single culture and the unknown dynamic nature of M. haemolytica shedding. Because these calves were commingled while awaiting the results of the culture, it is also possible that this change in prevalence could signify potential transmission. However, since our study was not designed to identify transmission, PFGE was not performed on the arrival screening cultures and therefore we are unable to provide evidence either for or against transmission of M. haemolytica in this study. Regardless of the exact cause, the change in apparent prevalence between screening cultures further emphasizes the potential problem with using a single culture at a single point in time or even 2 cultures interpreted in parallel to give an accurate assessment of M. haemolytica status.
There are many possible reasons why a single culture may not be sufficient to accurately represent the culture status of a calf even when samples are obtained and handled properly. First, when compared with the large surface area of a calf’s nasal passages, the surface area of a culture swab and the area it can sample are relatively small. Taking into account the potential diverse and dynamic microbial flora of the nasal mucosa of a calf, the potential for competitive inhibition, the unknown natural shedding pattern of M. haemolytica, and the variable rate of mucous production, it is not surprising that 1 culture swab cannot necessarily correctly classify a calf as negative.
Analysis by PFGE was instrumental in helping to characterize the clusters of M. haemolytica that were isolated, and allowed for the comparison of the similarity of the challenge strain to each selected isolate. Additionally, the PFGE results raised concerns about expecting a single culture to provide a true representation of the M. haemolytica population within the nasopharynx. The challenge strain used in this study was taken from a prior clinical case of BRD, had been utilized in another challenge study, 5 and, based on lesions observed during autopsy, was successful in inducing comparable pneumonic lesions. However, none of the isolates saved from the postchallenge NPCs matched the challenge strain, based on a 90% similarity threshold value, even though the challenge strain was placed in both the nasal cavity and the lung. Whether this was a result of the challenge strain not being an efficient colonizer in the presence of the other M. haemolytica types isolated from the NEG calves, not being present in the small area from which the swab was taken, or not being the single isolate selected from the colonies on the plate for PFGE analysis remains unclear.
The PFGE results from the POS calves showed that it is possible, when multiple isolates are taken from a single plate, to find more than 1 cluster of M. haemolytica. A preliminary report presented as a poster at the 2014 BRD Symposium (Taylor et al., Use of pulsed-field gel electrophoresis for characterization of Mannheimia haemolytica isolates from the upper and lower respiratory tracts of cattle, Poster presentation at the Bovine Respiratory Disease Symposium, 2014 July 30–31, Denver, Colorado) also indicated that more than 1 pulse-type of M. haemolytica can be found in an animal. Another study 7 found evidence that, based on plasmid profile, 2 or more strains of M. haemolytica can be found within a single calf. Current bacteriology practices depend on the assumption that if a single isolate is selected randomly from a plate it is likely to be the most predominant one on the culture plate and therefore the nasopharynx. 22 Although there may indeed be a “dominant strain” of M. haemolytica within a given sample, the selection of a single isolate is unlikely to be truly random and instead may suffer from unintentional selection bias due to potential differences in M. haemolytica colony morphology. Additionally, given the previously mentioned inherent limitations of NPCs, it may be unrealistic to extrapolate the results from the single isolate level to the entire nasopharynx. In addition to the multiple assumptions routinely made when performing standard bacterial culture techniques, 22 it is important to remember that the goal of routine diagnostic methods is not to identify diversity within a sample, but to separate colonies for further characterization such as antimicrobial susceptibility testing. Therefore, it is possible that routine diagnostic methods may not be optimal for detecting diverse strains of bacteria within the original sample. Furthermore, controversy exists regarding the correlation of the bacteria within the upper respiratory tract with the bacteria in the lung 8 especially at the individual calf level. 2 If paired culture and PFGE are to be used as diagnostic tools for M. haemolytica in future studies, it would be extremely beneficial to know how many isolates from each culture need to be subjected to PFGE to gain a true representation of the M. haemolytica strains present within each culture. Several statistical models involving Bayesian inference have been developed to estimate this number.3,11,22 However, to our knowledge, this approach has not yet been applied to M. haemolytica.
Other data collected during this pilot included CIS scores, rectal temperatures, and in the case of the challenged calves, lung scores, gross postmortem lesions, and lung cultures. In all phases, CIS and rectal temperature did not seem correlated with either NPC status or, where applicable, with lung lesions or lung cultures at autopsy. The negative results seen on the postmortem lung cultures could be a result of both the innate limitations of culture and the successful immune clearance of the bacteria by day 10. It is well established that rectal temperature and CIS are relatively insensitive and unspecific methods of diagnosing BRD 27 especially when compared to gross pulmonary examination at autopsy 17 or slaughter. Therefore, the lack of correlation between CIS, rectal temperatures, and autopsy findings seen in our study is not surprising and may be due to several confounding factors including individually housing the calves, high ambient temperatures, and the large amount of normal lung tissue left in each calf. Despite the presence of pulmonary lesions that were comparable to those seen in other challenge studies,5,17 these calves still had a large amount of normal lung tissue remaining and therefore may not have had appreciably abnormal respiratory character or behavior. Assignment of clinical illness scores may also have been confounded by individually housing the calves and thus changing their behavior and willingness to show clinical signs when under observation. Additionally, given the high ambient temperature during this trial, it was difficult to discern between mild signs of respiratory disease and mild signs of heat stress. These 3 factors, among others, collectively made it difficult to accurately judge clinical illness when compared with a more typical commercial setting.
Even though the sample size within each portion of our study was limited, the inconsistencies seen in culture status and the M. haemolytica isolates obtained suggest that caution should be used when interpreting the results of a single NPC and that extrapolation of the results to the entire nasopharynx or to the lower respiratory tract may not be possible. Furthermore, this study highlights the complex role of M. haemolytica in both healthy and ill animals and emphasizes the need for future research to further explore the epidemiology of M. haemolytica transmission.
Footnotes
Acknowledgements
We would like to thank Deepti Pillai for preparation of the challenge media, and David Amrine, Doug Shane, and Jeremy Kramer for helping manage cattle, samples, and data.
Authors’ contributions
BJ White and RL Larson contributed to conception and design of the study. SF Capik, DA Mosier, and RW Murray contributed to the design of the study. SF Capik, BJ White, RL Larson, and RW Murray contributed to acquisition, analysis, and interpretation of data. BV Lubbers and DA Mosier contributed to acquisition and interpretation of data. MD Apley contributed to interpretation of data. SF Capik and BJ White drafted the manuscript. All authors critically revised the manuscript, gave final approval, and agree to be accountable for all aspects of the work in ensuring that questions relating to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
a.
JorVet #J-272, Jorgensen Laboratories, Loveland, CO.
b.
Remel BactiSwab gel collection and transport system, Amies clear media; Thermo Fisher Scientific, Lenexa, KS.
c.
VetVu VFS-2B, Swiss Precision Products Inc., Spencer, MA.
d.
CASH dispatch kit, Accles & Shelvoke Ltd., Sutton Coldfield, West Midlands, United Kingdom.
e.
Bruker Daltonics Inc., Billerica, MA.
f.
Microscan turbidity meter, Dade-Behring, West Sacramento, CA.
g.
Sigma-Aldrich, St. Louis, MO.
h.
Bio-Rad Laboratories, Hercules, CA
i.
Life Technologies, Grand Island, NY.
j.
CHEF-Mapper XA system, Bio-Rad Laboratories, Hercules, CA
k.
SeaKem-Gold, Lonza Rockland Inc., Rockland, ME.
l.
Life Technologies, Grand Island, NY.
m.
VersaDoc imager, Bio-Rad Laboratories, Hercules, CA.
n.
Bionumerics v7.1, Applied Maths Inc., Sint-Martens-Latem, Belgium.
Declaration of conflicting interests
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: The authors, with the exception of RW Murray, declare that there are no conflicts of interest. RW Murray is employed by, and owns stock in, Zoetis Inc.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Funding was provided by Zoetis.