
Abstract
A real-time fluorescence resonance energy transfer polymerase chain reaction (qFRET PCR) coupled with melting curve analysis was developed for detection of Babesia canis vogeli and Hepatozoon canis infections in canine blood samples in a single tube assay. The target of the assay was a region within the 18S ribosomal RNA gene amplified in either species by a single pair of primers. Following amplification from the DNA of infected dog blood, a fluorescence melting curve analysis was done. The 2 species, B. canis vogeli and H. canis, could be detected and differentiated in infected dog blood samples (n = 37) with high sensitivity (100%). The detection limit for B. canis vogeli was 15 copies of a positive control plasmid, and for H. canis, it was 150 copies of a positive control plasmid. The assay could simultaneously distinguish the DNA of both parasites from the DNA of controls. Blood samples from 5 noninfected dogs were negative, indicating high specificity. Several samples can be run at the same time. The assay can reduce misdiagnosis and the time associated with microscopic examination, and is not prone to the carryover contamination associated with the agarose gel electrophoresis step of conventional PCR. In addition, this qFRET PCR method would be useful to accurately determine the range of endemic areas or to discover those areas where the 2 parasites co-circulate.
Pathogens transmitted by ticks of the genus Ixodes remain health problems in animals and human beings by causing babesiosis, ehrlichiosis, and hepatozoonosis. 10 Babesia species are apicomplexan parasites of erythrocytes and infect many species of mammals. 18 Three subspecies of Babesia canis (B. canis canis, B. canis rossi, and B. canis vogeli) have been recognized as the causative agents of canine babesiosis, a clinically significant and geographically widespread disease. 17 In particular, B. canis vogeli causes disease in domestic dogs and wild canids in southern Europe, the United States, Asia, and South Africa. 17 Rhipicephalus sanguineus (brown dog tick) is the main known vector of B. canis vogeli. 17
Hepatozoon canis is another apicomplexan protozoan parasite, and is the cause of canine hepatozoonosis, a disease that is prevalent in Asia, Africa, South America, southern Europe, and recently in the United States. 1 The tick, R. sanguineus, serves as a definitive host: Merogony occurs in the liver and spleen of the canine host; the host then becomes infected by ingestion of infected ticks. 1 Coinfections with both B. canis vogeli and H. canis have been reported. 14 In the past, laboratory diagnosis of these diseases was generally based on visual identification of parasites in erythrocytes. 2 Detection is difficult, especially in animals experiencing a chronic phase of infection or that have a low parasitemia, and false negatives are common.5,12 Polymerase chain reaction (PCR) provides a practical means to diagnose and differentiate infections with various Babesia and Hepatozoon spp. and has become a sensitive tool for evaluating treatment outcomes.3,11
Real-time PCR–based techniques for detection of canine Babesia spp. 19 or Hepatozoon spp.4,8 have been reported. The use of separate diagnostic assays for each pathogen is expensive and time consuming. In the current study, a single-tube real-time fluorescence resonance energy transfer (FRET) PCR, termed qFRET-PCR, using 1 fluorophore-labeled probe and 1 pair of primers to rapidly detect 2 pathogens, B. canis vogeli and H. canis, in dog blood samples, is described.
A total of 54 dog blood samples were used. Of these, 21 were infected with B. canis vogeli, 16 with H. canis, 1 with mixed B. canis vogeli and H. canis, 5 with Ehrlichia canis, and 1 with Anaplasma platys. Pathogens in all of these samples were identified by microscopic examination of Giemsa-stained blood films followed by PCR and Sanger sequencing 7 or pyrosequencing. 6 Brugia pahangi and Dirofilaria immitis, each found in 5 samples, were identified during microscopic examination of Giemsa-stained blood films. The ethylenediamine tetra-acetic acid–treated blood samples were obtained from dogs treated at the Private Animal Hospital, Bangkok and from stray dogs in Khon Kaen Province, Thailand. All specimens were blind-labeled. The GenBank accession numbers for sequences from B. canis vogeli (n = 21) are KF621061 to KF621081. For H. canis (n = 16), the GenBank accession numbers are KF621082 to KF621097. In addition, DNA from human blood samples positive for Plasmodium falciparum (n = 2), human white blood cells (n = 1), and healthy dog blood samples (n = 5), all obtained from the frozen sample bank at the Department of Parasitology, Faculty of Medicine, Khon Kaen University, were included as controls. The study protocol was approved by the Animal Ethics Committee of Khon Kaen University, based on the Ethics of Animal Experimentation of the National Research Council of Thailand (reference no. 0514.1.12.2/50).
For each blood specimen, 200 µL was placed into a 1.5-mL microcentrifuge tube, homogenized using a disposable polypropylene pestle, a and DNA extracted using a commercial kit. b The DNA samples were eluted in 50 µL of 5 mM Tris-HCl (pH 8.5), of which 5 µL were used in the qFRET-PCR and conventional PCR reactions.
A commercial system c was used for amplification and quantification. The qFRET PCR was performed in glass capillaries. The method used 1 pair of specific primers, a forward primer (Ba18S_For: 5′-GCGATGGACCATTCAAGT-3′) and reverse primer (Hepa18S_Rev: 5′-CTTGCCCTCCAATTGATACTT-3′). These primers were designed from an alignment of the partial sequences of B. canis vogeli and H. canis 18S ribosomal RNA genes (GenBank accession nos. HQ662635 and AF176835, respectively) and were predicted to amplify products of 267 and 278 base pairs (bp) respectively. For detection of amplification, a commercial kit d was used as recommended by the manufacturer. Briefly, a pair of adjacent oligoprobes was hybridized with the amplicons generated by Ba18S_For and Hepa18S_Rev. One probe was labeled at the 5′-end (Hepa18S_LC705: 5′-Red 705-GCGCGCAAATTACCCAATTCTAACAGTTTGAGAG-Phosphate-3′) and the other labeled at the 3′-end (Hepa18S_FL: 5′-GAAACGGCTACCACATCTAAGGAAGGCAGC-Fluo 530-3′). e Probes and primers were designed using a commercial software package f and were synthesized commercially.e,g A schematic diagram of the hybridization analysis of the primers and probes is shown in Figure 1. When the probes were hybridized to the same DNA strand internal to the PCR primers, the probes came in close proximity and produced a FRET. The PCR mixture contained 1× commercial DNA master mix, d 2 mM MgCl2, 0.3 µM Ba18S_For, 0.3 µM Hepa18S_Rev, 0.2 µM Hepa18S_FL, and 0.2 µM Hepa18S_LC705. The total reaction volume was 20 µL. A commercial qPCR machine and detection system h was used for amplification and quantification. The samples were pre-incubated for 10 min at 95°C, then run through 45 cycles of denaturation (10 sec at 95°C), annealing (30 sec at 55°C), and extension (15 sec at 72°C). After amplification, a melting curve was produced by heating the product at 20°C/sec to 95°C, cooling it to 59°C, keeping it at 59°C for 10 sec and then slowly heating it at 0.2°C/sec to 80°C. The fluorescence intensity change was measured throughout the slow heating phase. Each run contained at least 1 negative control consisting of 5 µl of distilled water. For improved visualization of the melting temperatures (Tm) and melting peaks, the analyses were done as recommended by the manufacturer of the qPCR machine. h The length of each PCR product was determined by electrophoresis in a 2% agarose gel.
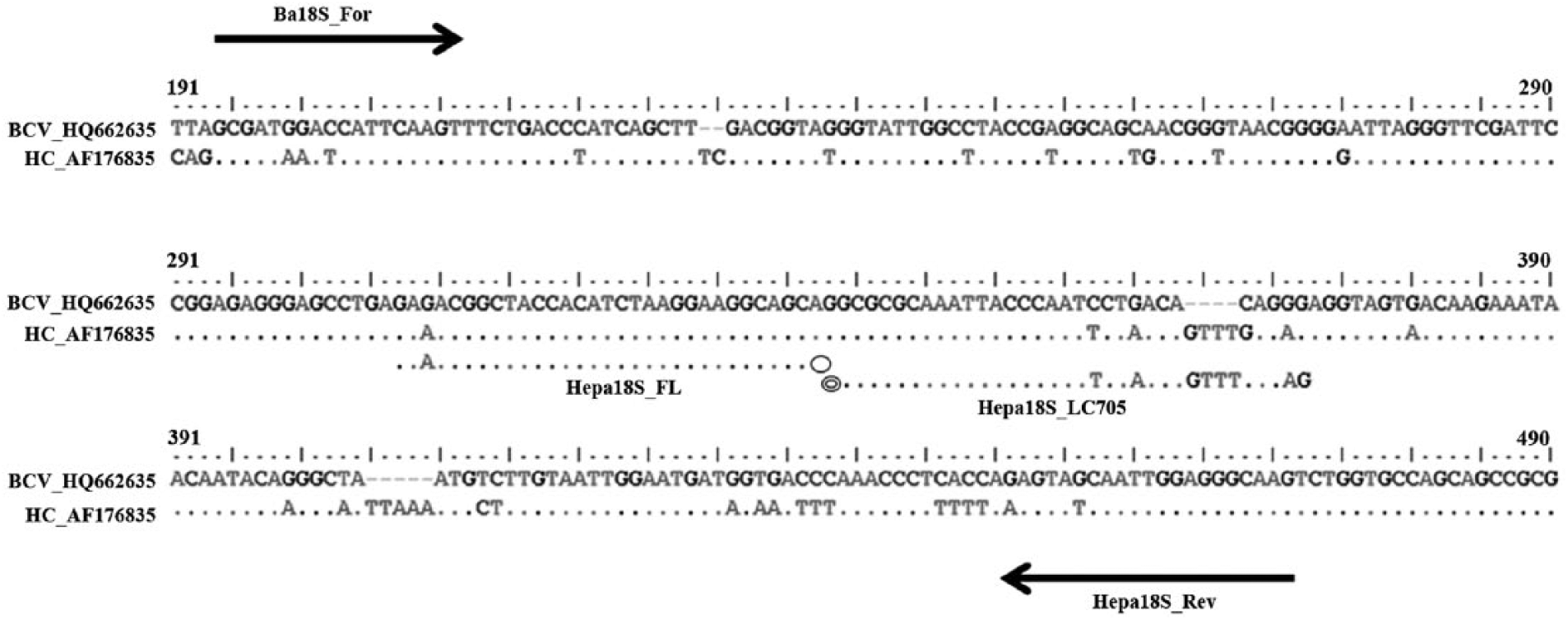
Schematic illustration of the hybridization analysis of the primers (arrows indicate the position of Ba18S_For and Hepa18S_Rev primers) and anchor and detection probes for the 18S ribosomal RNA gene from Babesia canis vogeli and Hepatozoon canis. The probe Hepa18S_FL was labeled with fluorescein at the 3′-end and served as anchor probe for the sensor Hepa18S_LC705 probe that was labeled with a fluorophore at the 5′-end. Oval indicates fluorescein; double oval indicates fluorophore.
A positive control plasmid of each parasite was constructed by cloning a PCR product from B. canis vogeli or from H. canis into a commercial vector i according to the protocol of the manufacturer. The PCR products were obtained using the Ba18S_For and Hepa18S_Rev primers for amplification of either B. canis vogeli or H. canis DNA from an appropriate blood sample. The PCR reaction consisted of a 25-µL reaction volume containing 1× PCR buffer j with 0.2 mM of each deoxynucleotide, 2 mM MgSO4, 0.2 µM of each primer, 0.625 U of high fidelity Taq DNA polymerase, j and 2 µL of the DNA template. The amplification procedure was as follows: 5 min at 94°C for initial denaturation followed by 35 cycles of denaturation for 30 sec at 95°C, annealing for 30 sec at 55°C, and extension for 30 sec at 72°C, followed by a final extension for 10 min at 72°C. Each PCR product was cut from the gel, purified, and then ligated into the plasmid. The plasmids were propagated in Escherichia coli, and the inserts were sequenced in both directions to confirm their identities. The resulting sequences were identical with the sequences in the GenBank database for B. canis vogeli and H. canis.
The sensitivity of the qFRET-PCR was determined using 5 µl of serial 10-fold dilutions of either B. canis vogeli– or H. canis–positive control plasmids (1.5 × 101–1.5 × 109 copies/reaction) in water. The limit of detection for the B. canis vogeli target DNA region was 15 copies of the positive control plasmid (Fig. 2A), whereas for H. canis, it was 150 copies of the positive control plasmid (Fig. 2B), when using a cutoff detection limit of 40 cycles. No fluorescence signal was detected when purified DNA from E. canis, A. platys, B. pahangi, D. immitis, or P. falciparum, and the noninfected dog blood samples (n = 5) and human white blood cells (n = 1) were tested (Fig. 3A). The qFRET-PCR successfully amplified the predicted 267-bp product from the DNA of all 21 B. canis vogeli–infected samples and the predicted 278-bp product from the DNA of all 16 H. canis–infected samples (typical results are shown in Supplementary Fig. 1; available at jvdi.sagepub.com/supplemental). The only sample known to be infected with B. canis vogeli and H. canis gave 2 melting peaks (Fig. 3A), indicating that the method can be used for detecting coinfections involving both pathogens.
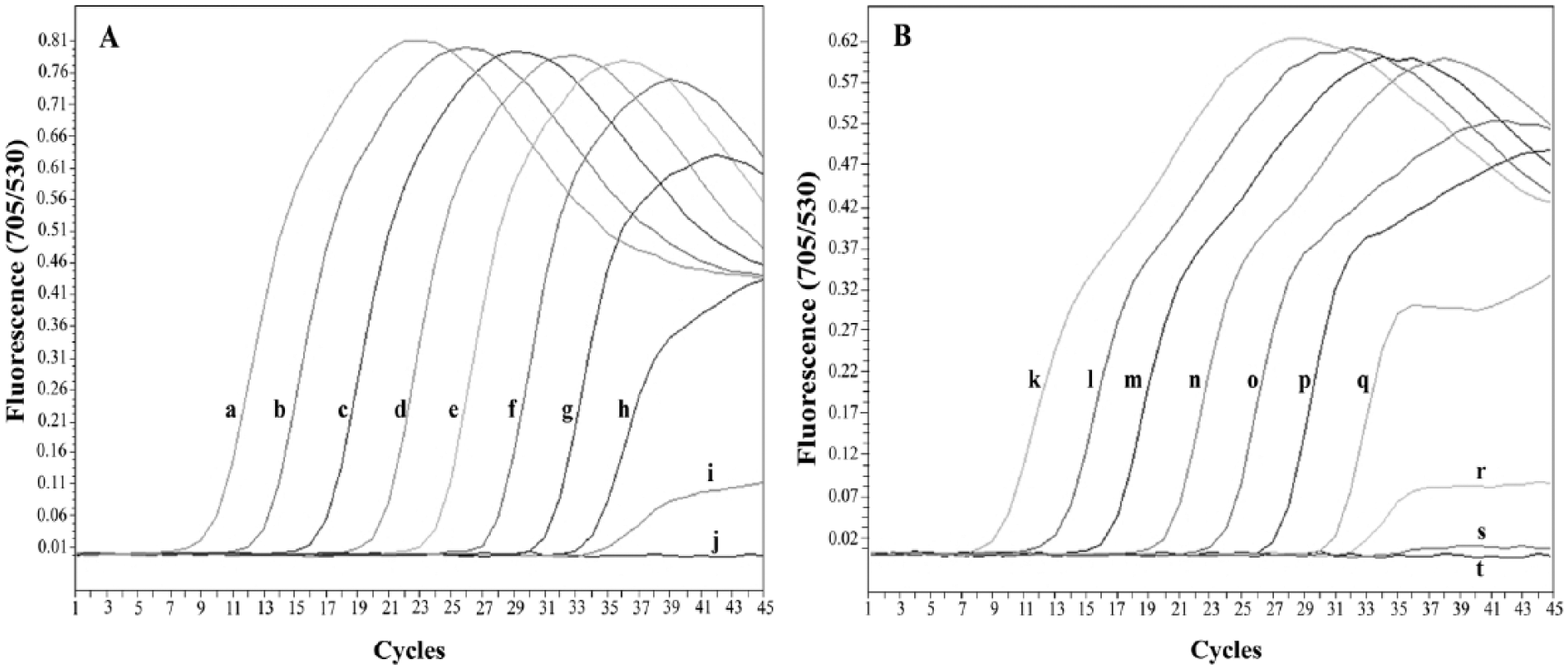
Amplification curves show the analytical sensitivity of real-time fluorescence resonance energy transfer polymerase chain reaction assay for detecting Babesia canis vogeli (
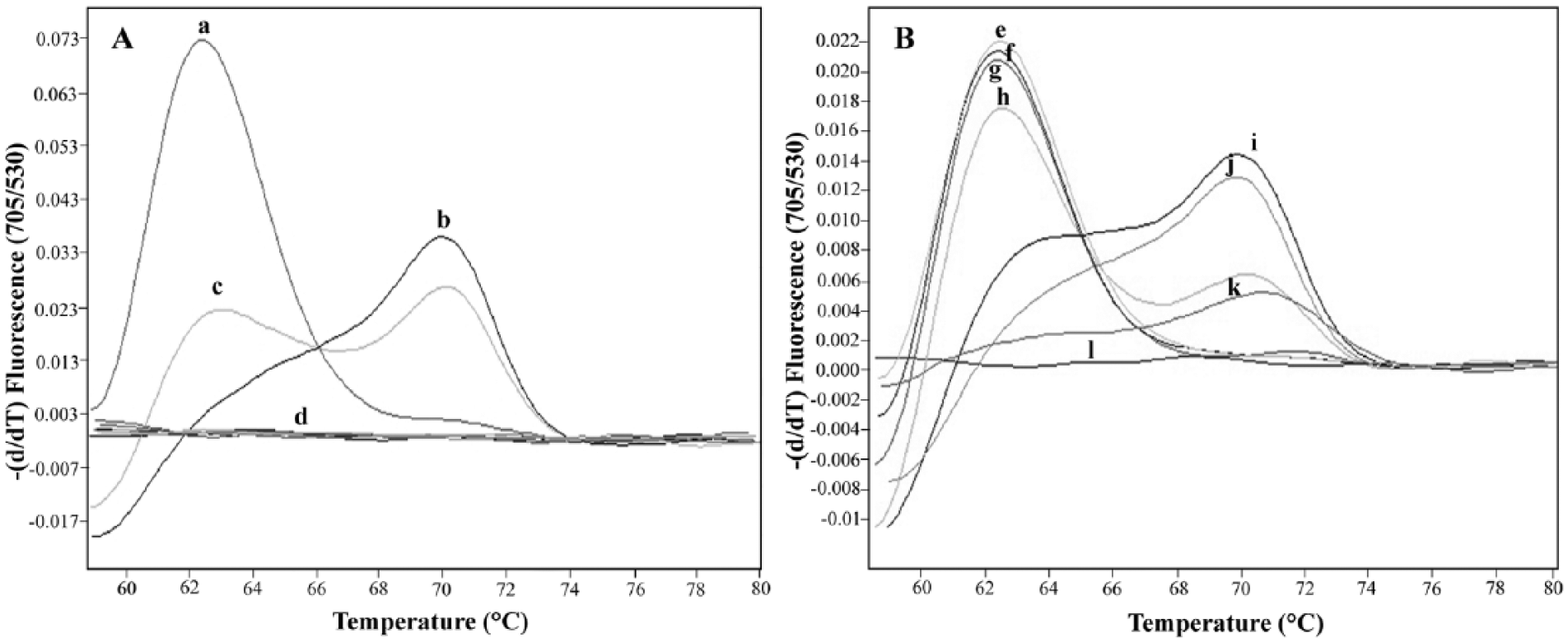
Representative melting curve analyses of 1 fluorophore-labeled probe hybridized to the amplification products of Babesia canis vogeli and Hepatozoon canis 18S ribosomal RNA genes.
Typical melting curve analyses are shown in Figure 3B. The mean (± the standard deviation) of Tm values of B. canis vogeli– and H. canis–infected dog blood samples were 62.71 ± 0.35 (n = 21) and 70.89 ± 0.66 (n = 16), respectively. No Tm value could be detected for the negative blood samples or the other control DNA samples. Hence, the qFRET-PCR showed 100% sensitivity and specificity.
Babesia and Hepatozoon species cause important tick-borne protozoal diseases of dogs. The diagnosis of canine babesiosis and hepatozoonosis by detection of pathogens in peripheral blood under a microscope is labor intensive and time consuming, with low sensitivities.5,12 A PCR has been previously reported for detection of Babesia spp. 11 and Hepatozoon spp. 3 as well as simultaneous detection of both agents in canine blood samples using 1 primer pair. 13 However, the PCR methods require agarose gel electrophoresis for analysis of amplicon size, are time consuming, and are prone to the potential risk of crossover contamination. 16 The qPCR assay has progressively superseded conventional PCR in diagnosis for infectious diseases because of its greatly improved efficacy of molecular detection. 8 In general, qPCR methods are not only accurate, rapid, and can measure the specific DNA quantity in samples, but also allow for differentiating species or strains of several pathogenic agents by melting curve analysis. 9 Furthermore, qPCR methods offer high throughput. 9 A qFRET-PCR assay for the simultaneous detection of Babesia and Hepatozoon in a single tube assay, however, has not, to the authors’ knowledge, previously been reported. In the current study, a qFRET-PCR coupled with melting peak analysis was developed for the detection of B. canis vogeli and H. canis in dog blood. The method gave high sensitivity, shown by the fact that the assay could detect at least 15–150 copies per reaction of positive control plasmids and that all 38 infected dog blood samples that were positive by microscopy were also positive in the qFRET-PCR. No fluorescence signal was detected for the nontarget control DNA samples or the blood samples from the 5 noninfected dogs, indicating high specificity.
Based on a bioinformatic analysis, the primers and the probes developed in the current study can all hybridize with nucleotide sequences in the GenBank database of B. canis canis (KF499115), B. canis rossi (JQ613105), and Hepatozoon americanum (AF176836), but not with Babesia gibsoni. Hence, the qFRET-PCR method developed in this study cannot differentiate between B. canis vogeli, B. canis canis, and B. canis rossi or between H. canis and H. americanum. This problem does not affect the treatment outcome in babesiosis-infected dogs, as the treatment of canine babesiosis is quite similar, regardless of causative species. 15 However, treatment of canine hepatozoonosis is different between H. canis and H. americanum, as the infected target cells and/or organs are different. 1
In conclusion, the qFRET-PCR approach with melting curve analysis developed in the current study overcomes the requirement for laborious microscopic examination and allows many samples to be processed at the same time. The assay could be useful as an alternative method of studying the epidemiology of canine babesiosis and hepatozoonosis as well as for prevention and control of both parasites in regions where both agents are circulating.
Footnotes
Acknowledgements
The authors wish to acknowledge the support of the Khon Kaen University Publication Clinic, Research and Technology Transfer Affairs, Khon Kaen University, for their assistance. The authors also thank Prof. David Blair for valuable suggestions.
a.
Polypropylene pestles, Bellco Glass Inc., Vineland, NJ.
b.
Nucleospin tissue kit, Macherey-Nagel GmbH & Co., Düren, Germany.
c.
Research products, Roche Applied Science, Mannheim, Germany.
d.
LightCycler FastStart DNA Master HybProbe kit, Roche Applied Science, Mannheim, Germany.
e.
TIB MOLBIOL GmbH, Berlin, Germany.
f.
The LC probe design software, Roche Applied Science, Mannheim, Germany.
g.
Sigma-Aldrich Pte. Ltd., Singapore.
h.
LightCycler 2.0 instrument operator’s manual, Roche Applied Science, Mannheim, Germany.
i.
pGEM-T easy vector, Promega Corp., Madison, WI.
j.
Roche Applied Science, Mannheim, Germany.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This research was funded by grants from the National Science and Technology Development Agency (Discovery Based Development Grant); the Higher Education Research Promotion and National Research University Project of Thailand, Office of the Higher Education Commission through the Health Cluster (SHeP-GMS) Thailand, the Faculty of Medicine, Khon Kaen University (TR57201), and the TRF Senior Research Scholar Grant, Thailand Research Fund (RTA5580004).