
Abstract
New World screwworms, Cochliomyia hominivorax (Coquerel, 1858), were once devastating pests of warm-blooded animals in the United States before they were successfully eradicated using the sterile insect technique. Guarding against the introduction of screwworms to North America or any other screwworm-free area relies on rapid, reliable identification of suspected cases. In the current study, the DNA from excised markers generated by randomly amplified polymorphic DNA polymerase chain reaction was used as the basis to generate 2 species-specific sequence-characterized amplified region molecular markers. Resulting primer pairs, named CR92A1 and J1A2 (each with forward and reverse components), produced amplicons of 852 and 848 base pairs, respectively. The 2 primer pairs successfully discriminated between C. hominivorax, Cochliomyia macellaria (Fabricius, 1775), 8 other species of blowflies, 3 noncalliphorid dipterans, and 1 nondipteran outlier. These primers may become important tools for veterinary laboratories and the screwworm eradication and exclusion program for rapid identification or verification of suspicious larval samples in presumed outbreaks.
Keywords
Introduction
The use of the sterile insect technique in an eradication program throughout the United States, Mexico, and Central America has eliminated the screwworm, Cochliomyia hominivorax (Coquerel, 1858; order Diptera, family Calliphoridae), from these regions, providing relief to livestock producers from annual economic losses exceeding $1.5 billion. 31 A barrier zone against reintroduction has been established at the Darien Gap in Panama. 20 South of this zone, the screwworm continues to be an economic pest causing annual profit losses estimated to exceed $3.5 billion for livestock producers, wildlife, and human beings in South America.23,31
Aside from the economic havoc caused by the screwworm where it is endemic, the possibility of reintroduction where previously eradicated or introduction to nonendemic areas poses a threat worldwide. The global community inadvertently could spread the screwworm outside its geographical range by human travel, international livestock trade and movements, and transport of pets and service animals. For example, the introduction, discovery, and, in some instances, necessary eradication of the screwworm has been documented in Libya in 1988,3,21 France in 1989, 5 Australia in 1992, 26 and Mexico in 1992 and 1993. 10 Cochliomyia hominivorax outbreak prevention is dependent on effective and reliable monitoring, enforcement of strict quarantine measures, and the swift implementation of a control program when necessary (USDA-APHIS: 2013, Disease response strategy: New World screwworm myiasis. Available at: http://www.aphis.usda.gov/animal_health/emergency_management/downloads/nws_myiasis_disease_strategy.pdf). To achieve this, accurate and timely species identification is imperative. Many facultative myiasis-causing and scavenger flies, which are very difficult to identify in the early life-stages, are also attracted to wounds; this includes the secondary screwworm, Cochliomyia macellaria (Fabricius, 1775), a morphologically similar species that was not taxonomically distinguished from screwworm until 1933.6,12 It is also important that these identification methods have the potential to be adapted for clinical and field use.
Molecular biology techniques provide tools to discriminate insect species, including the screwworm. One of the earliest reported techniques to successfully distinguish screwworms was polymerase chain reaction–restriction fragment length polymorphism of mitochondrial DNA.7,18,29 Randomly amplified polymorphic DNA polymerase chain reaction (RAPD-PCR) was used later and found to be useful and quick in discriminating C. hominivorax from C. macellaria. 28 Using RAPD-PCR, however, has potential problems with reproducibility in different laboratories. Amplified fragment length polymorphism 1 (AFLP) has been used to differentiate between C. hominivorax and C. macellaria, but this is a complex technique. 15 As an alternative to the molecular techniques, an enzyme-linked immunosorbent assay (ELISA) has been developed to identify C. hominivorax. 9 However, a major drawback to the ELISA is that insects must either be kept alive, on ice, or frozen immediately to avoid deterioration prior to testing. 19 The purpose of the current study was to develop and use, from existing RAPD-PCR markers, sequence-characterized amplified region (SCAR) markers and test the hypothesis that SCAR would differentiate between C. hominivorax, C. macellaria, other calliphorids, and noncalliphorids.
Materials and methods
Fly samples and DNA extraction
Archived specimens of C. hominivorax and C. macellaria (identification verified by SR Skoda, 1991–1995, using characters from published keys [Spradbery JP: 2002, A manual for the diagnosis of screw-worm fly. CSIRO Division of Entomology, Canberra, Australia. Available at: http://www.animalhealthaustralia.com.au/wp-content/uploads/2011/04/Manual-for-the-Diagnosis-of-Screw-Worm-Fly.pdf], and stored at −80°C) were used in the current study. The samples contained both wild types and laboratory-reared specimens in both larval and adult stages.
Frozen fly specimens from an earlier project, 25 including 8 calliphorids: Lucilia illustris (Meigen, 1826), Lucilia sericata (Meigen, 1826), Phormia regina (Meigen, 1826), Cynomya cadaverina (Robineau-Desvoidy, 1830), Calliphora vicina (Robineau-Desvoidy, 1830), Chrysomya rufifacies (Macquart, 1843), Lucilia coeruleiviridis (Macquart, 1855), and Bufolucilia silvarum (Meigen, 1826; syn. Lucilia silvarum); 3 noncalliphorids: Ophyra sp., Musca domestica (Linnaeus, 1758), Stomoxys calcitrans (Linnaeus, 1758); and 1 nondipteran outlier: Spodoptera frugiperda (Smith, 1797), were used for extraction of DNA using a phenol–chloroform method as described in an earlier project. 25 Whole insects and extracted DNA were stored at −80°C. Prior to use for the present study, the DNA was quality-checked on 1.5% agarose gels and quantified using a spectrophotometer. a The DNA concentrations were approximately100 ng/µl.
Additional fly collections were made (L. illustris, L. sericata, P. regina, C. cadaverina, B. silvarum, C. vicina, and S. calcitrans), and DNA was extracted as described above. These newly collected samples were used to confirm preliminary results. DNA was extracted from C. hominivorax and C. macellaria and stored. The DNA concentration of these samples, determined by the use of a spectrophotometer, a was also approximately 100 ng/µl.
Quick frozen larval and adult samples were washed once with autoclaved double-distilled water for 10 min. Larvae stored in 95% ethanol were washed twice (15 min each wash) with autoclaved double-distilled water. The gut was removed from both larval and adult samples. In addition, the legs, wings, and head of the adult specimens were removed and retained as vouchers at −80°C storage in the Department of Entomology Genetics Laboratory at the University of Nebraska (Lincoln, Nebraska). Individual specimens were placed in an autoclaved 1.5-ml microcentrifuge tube (one tube for each sample). Extraction procedures were as previously described,1,27,28 and the extracted DNA was stored at 4°C for short term and −20°C for long term.
Randomly amplified polymorphic DNA polymerase chain reaction
The RAPD-PCR assays were performed using a commercial system b and commercial reagents. b The DNA template for the PCR was diluted to a concentration of approximately 2.5 ng/µl. The RAPD-PCR solutions and temperature profile followed the previously established protocol.27,28 The random primer named OPA-12 (TCGGCGATAG) c was used because it showed consistent and reproducible banding patterns in previous studies.27,28 In replicating the RAPD-PCR, DNA marker bands that discriminate the species were reproduced at approximately 860 bp for C. hominivorax and approximately 292 bp for C. macellaria using primer OPA-12 (Fig. 1).
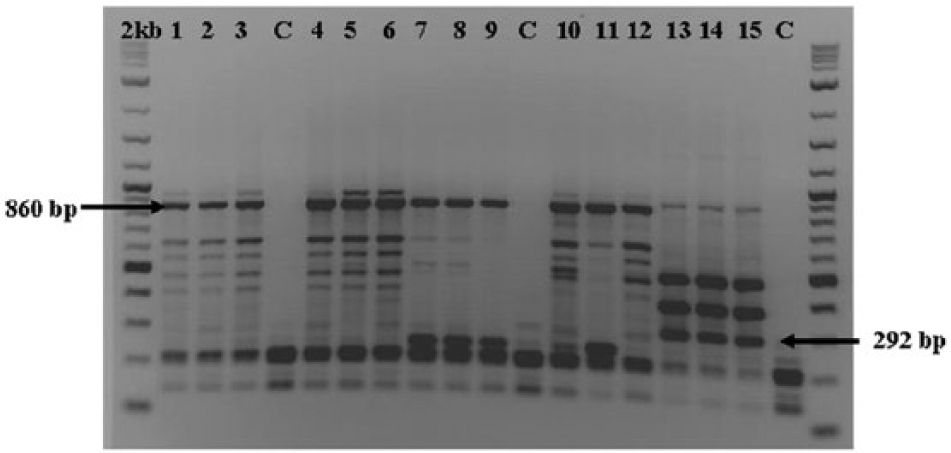
Randomly amplified polymorphic DNA profiles from the genomic DNA of Cochliomyia hominivorax (lanes 1–12) and Cochliomyia macellaria (lanes 13–15) adults and larvae. Side-by-side comparison using primer OPA-12. Lanes 1–3: Jamaica (J1); lanes 4–6: Panama (P95); lanes 7–9: Mexico (PA34); lanes 10–12: Costa Rica (CR92); lanes 13–15: Nebraska (CMN); C = control; 2 kb = 2-log DNA ladder. e Arrows indicate diagnostic bands approximately 292 bp and approximately 860 bp.
DNA purification and primer design for developing sequence-characterized amplified regions
The approximately 860-bp marker bands (C. hominivorax; representing 11 individuals from Mexico, Costa Rica, Panama, and Jamaica) and the approximately 292-bp marker bands (C. macellaria; representing 6 individuals from Nebraska and Jamaica) of individual specimens generated with RAPD-PCR using primer OPA-12 were excised from the agarose gel and then purified using a commercial kit. c The pure DNA was then eluted with double-distilled nuclease-free water.
The PCR amplicon was inserted into a commercial cloning vector a to transform Escherichia coli following the supplier’s protocol. After transformation, the bacteria plasmid DNA was purified using a commercial kit. c Restriction digest was performed to screen and provide visible proof of desired results. The plasmid DNA (from 13 colonies) was then sent for sequencing using T3 and T7 primers to the University of Nebraska DNA Sequencing Facility (Lincoln, NE).
After gaining sequencing results (from 10 of the 13 colonies), the inserted vector sequences were easily identified and isolated using a freeware program (CAP3). 13 The EcoRI sites flanking the vector insertion site were located. That restriction site plus the bases leading up to the insertion site were removed leaving the insertion sequence of both T3 and T7 primers. The modified sequences of both the T3 and T7 sequences (with vector and EcoRI sites removed) were put into FASTA format (GenBank accession nos. KJ740479–KJ740488), copied and pasted into CAP 3, and the contig assembled the entire insertion sequence generated by the T3 and T7 primers.
After the contig assembly, a freeware program (ClustalW17,30) was used to align the sequences of C. hominivorax. The optimal primer picks indicated by the freeware program Primer324 were then chosen for both forward and reverse primers that were named CR92A1 and J1A2 (Table 1). Selection factors included closeness of melting temperatures for the primers, percentage of GC content (50% or more), primer length (optimum 20 bp), and the possible generation of primer-dimer or 3′ self-complementary strand generation. Two sets of primers, CR92A1 and J1A2 with forward and reverse in each set, were then ordered. d Primer concentrations aliquots of 10 µM were made.
Primers and melting temperatures used in the current study.

Developing PCR of sequence-characterized amplified regions
Reagents were obtained from a commercial source b unless otherwise indicated. The master mix for each reaction consisted of 15.5 µl of nuclease-free water, 2.5 µl of 10× PCR buffer, 1.5 µl of 25 mM MgCl, 1.25 µl each of the forward and reverse primers d (either CR92A1 or J1A2), 1.0 µl of 10 mM deoxynucleotide triphosphates, and 1.0 µl of Taq b polymerase for a total of 24 µl. One µl of DNA template (100 ng/µl) was then added to each tube. A negative control of PCR master mix (25 µl) without DNA template was included to check for primer-dimer and contamination.
The PCR temperature profile selected after optimization (variation of number of cycles and temperatures used) for the 2 primers consisted of: an initial denaturalization at 94°C for 5 min, 30 cycles at 94°C for 30 sec, 56°C for 45 sec, and 72°C for 60 sec. An additional 10-min extension step was done at 72°C followed by a 4°C infinite soak. Samples were stored at −20°C. The PCR products (10-µl aliquot) were subjected to gel electrophoresis (1.5% gel), in the presence of ethidium bromide (approximately 0.5 µg/ml), at 80 V for 3 hr. A 2-µl volume of a commercial 2 kb-log (1,000 µl/ml) molecular weight standard c was added to additional wells of all sample sets for comparison purposes. Negative controls were used with each sample set.
Results
Two distinct RAPD-PCR molecular markers of lengths 852 bp and 848 bp were generated, isolated, and identified from amplicons originally generated using RAPD-PCR with the OPA-12 primer (Fig. 1). The resultant SCAR primers (Table 1) could reliably identify C. hominivorax regardless of stage of development (Fig. 2). A BLAST (http://www.ncbi.nlm.nih.gov/blast/Blast.cgi) search showed no similarities to sequences on file in the GenBank database. The 852- and 848-bp contigs developed from resultant sequences (GenBank accession numbers KJ740479–KJ740488) contained part of the OPA-12 primer, and the sequence did not match the cloning vector. No sequence information was retrieved from the approximately 292-bp RAPD-PCR marker band from C. macellaria.
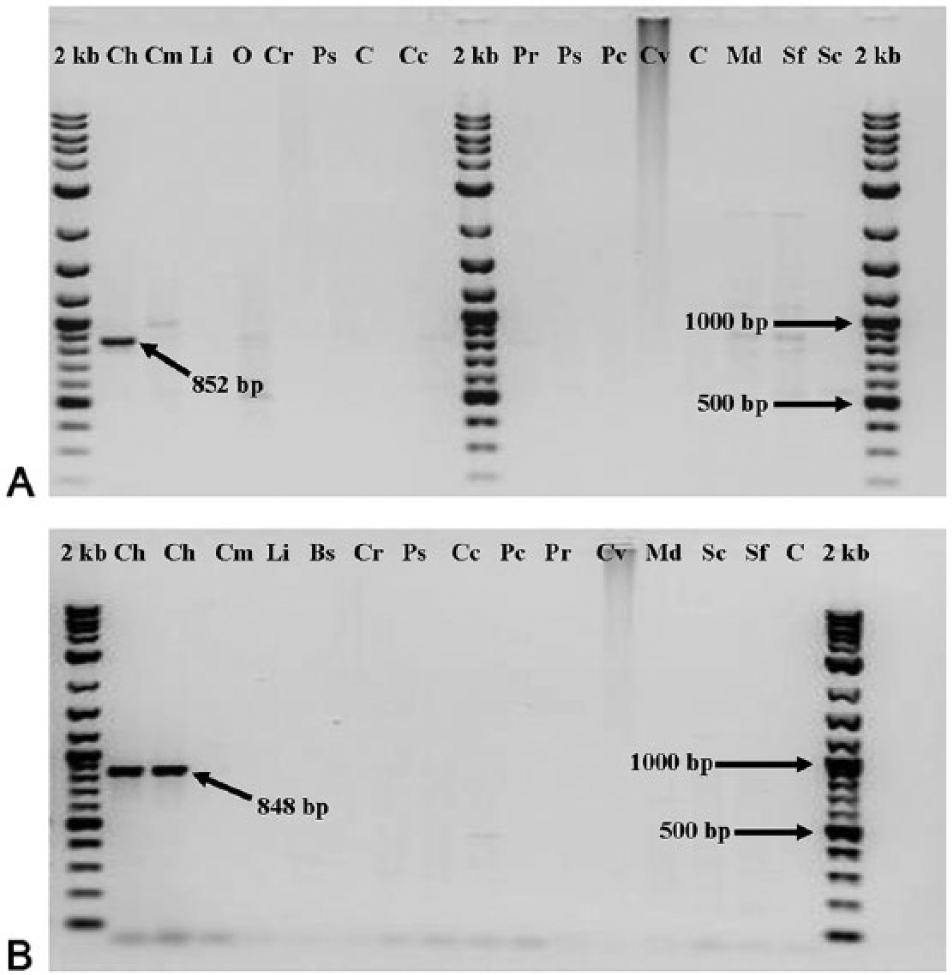
Sequence-characterized amplified region profiles from the genomic DNA of multiple adult flies (set 1) with CR92A1 primer (
The appearance of the SCAR marker appears to be a random event without regard to origin of the specimen. The 852-bp sequence was found in all 4 of the geographic origins of specimens—Mexico, Panama, Costa Rica, and Jamaica. The 848-bp sequence was found in 2 of the 4 locations—Mexico and Jamaica—yet the resultant primer (J1A2) was successful in identifying all screwworm samples. It is possible that sequencing more colonies and/or individuals would yield more of the latter sequence.
The main goal of the current work was to use RAPD-PCR to generate SCAR-PCR markers that were more reliable, thus eliminating potential reproducibility problems associated with RAPD-PCR. Both primers, CR92A1 and J1A2, amplified DNA from C. hominivorax (Fig. 2). The primer pair CR92A1 showed the presence of some irregular, faint banding patterns with other flies. After the final optimization of primers CR92A1, the J1A2 primers were evaluated. The primer pair generated from J1A2 showed a total absence of bands when used with DNA from C. macellaria (Fig. 2). Subsequent trials with multiple species of flies with both primers showed the same positive identification of C. hominivorax and negative results for other calliphorids, noncalliphorids, and the nondipteran (Fig. 2). Secondary confirmation tests were carried out using only primer J1A2 and resulted in the same positive identification of C. hominivorax and negative results for other calliphorids, non-calliphorids, and the nondipteran, S. frugiperda.
Discussion
Accurate and rapid identification of C. hominivorax is an important part of outbreak prevention and quarantine enforcement. Because of the difficulties using morphological identification (time-consuming and very difficult with first instars), 6 molecular techniques provide tools to augment timely and accurate identification. Using RAPD-PCR generates many genetic polymorphisms valuable in molecular genetics research and for sample identification.27,28 However, the potential lack of reproducibility has led to a search for more reliable and reproducible techniques when the question pertains to outbreak situations (with potential high cost of containment and eradication). The use of RAPD-PCR primer OPA-12 to generate species-specific bands and developing SCAR markers that are allele-specific eliminated the problems with RAPD-PCR reproducibility. Because SCAR-PCR uses longer primers specific to the target sequence, amplification is under stringent conditions and can be used on dead, frozen, or alcohol-preserved specimens. Both primer sets developed in the current work, CR92A1 and J1A2, differentiated between C. hominivorax, C. macellaria, other calliphorids, noncalliphorids, and a nondipteran. The primer J1A2 showed an especially pristine, clear gel with minimal amplification of DNA from the other insect samples. When using these primers, it is recommended that a negative (no DNA) and positive control (DNA of known quality from C. hominivorax) be used as well as an internal amplification control, such as primers with similar amplification temperature profiles that amplify mitochondrial DNA targets of a different size 29 (i.e., primers named Sp-1 and Sp-2; Pornkulwat S: 1998, Molecular genetic markers among and within populations of the European corn borer and screwworms, 143 pp. PhD dissertation, University of Nebraska, Lincoln, NE).
Many different molecular techniques have been used to generate SCAR markers. A SCAR useful in identifying apple scab resistance has been developed from AFLP markers. 32 A 255-bp SCAR was developed from a PCR product of porcine genomic DNA. 8 The SCARs developed from RAPD-PCR markers were used to discriminate between crocodilian species 33 as well as to rapidly identify biotypes of the whitefly, Bemisia tabaci (Gennadius, 1889). 16 Intersimple sequence repeat fingerprints were used to develop SCARs for detecting toxic phytoplankton. 4 Development of primers that provide a high degree of confidence for identification of species is of interest under many circumstances (e.g., import and export inspections, and drug resistance). Specifically, the primers developed in the current study are now available to determine if suspicious larval samples from animal wounds are New World screwworms, aiding quarantine officials in protecting against invasion to regions free of this major pest.
Some SCAR markers have been developed that are associated with the sex chromosomes in various organisms. The DNA sequence OmyP9 amplifies preferentially in male rainbow trout, Oncorhynchus mykiss (Walbaum, 1792), suggesting an association with the Y chromosome. 14 The SCARs associated with female willow shrub, Salix eriocephala (Michx.), have also been developed. 11 Future work in this area with screwworms could be useful in completing screwworm genetic “maps.”
Some RAPD-PCR markers have been used to generate molecular markers linked to the dominant Beta (B) locus that increases lycopene in tomatoes. 34 These B markers are useful in marker-assisted selection breeding. Molecular marker(s) linked to a trait of interest in the screwworm would allow true breeding genotypes to be identified. This could be a valuable tool in the maintenance and monitoring of mass-produced screwworm colonies used for eradication efforts employing the sterile insect technique.
Although the use of polyacrylamide gels to resolve and isolate RAPD bands 2 is usually recommended because of poor resolution of comigrating bands in agarose gels, 22 the relatively comparable size of the 2 cloned fragments (a difference of only 4 bp) would most likely prove impossible to separate without cloning. However, for future work, the use of acrylamide gel with the initial RAPD product could identify other bands of interest that could prove more helpful in intraspecific differentiation of C. hominivorax.
The SCAR markers in the current study, similar to the RAPD bands from which they were developed, are dominant markers that generate a single condition of presence or absence. Although the SCAR marker also has the potential to be developed as a codominant marker, often the monomorphic SCAR product results in the loss of initial polymorphism shown in the RAPD. 34 Recovery of the polymorphisms identified by the RAPD technique can be accomplished by further optimization of the PCR parameters or redesigning the primers. Another method would be to use restriction enzyme digestion of the SCAR-amplified products to develop cleaved amplified polymorphic sequences. 34 The codominant and allele-specific SCARs could then be used to accurately and quickly identify homozygous recessive, heterozygous, and homozygous dominant genotypes and may be useful in determining geographical origin of specimens in the future 34 ; this ability would be valuable in the investigation of screwworm outbreaks.
Overall, SCAR markers are superior to previously used molecular techniques (AFLP, RAPD, restriction fragment length polymorphisms, ELISAs) for the positive identification of screwworm. Therefore, the hypothesis that DNA bands (SCARs) determined from sequencing common RAPD marker bands among C. hominivorax can be used to differentiate interspecific blowfly populations has been confirmed.
Footnotes
Acknowledgements
The authors acknowledge S. Upeka Samarakoon, John Wang, Jayne Christen, and Maureen Gorman for their assistance with this work. This work was done in cooperation with the Institute of Agriculture and Natural Resources, University of Nebraska, Lincoln, Nebraska.
Authors’ note
Mention of a proprietary product does not constitute endorsement or recommendation for its use by the U.S. Department of Agriculture.
a.
NanoDrop UV-visible spectrophotometer, TOPO-4 cloning vector; Thermo Fisher Scientific Inc., Waltham, MA.
b.
Perkin-Elmer GeneAmp PCR system 9700, polymerase chain reaction reagents (i.e. Stoffel fragment Taq polymerase for RAPD, AmpliTaq Gold DNA polymerase for SCAR, buffers, etc.); Applied Biosystems, Foster City, CA.
c.
MinElute gel extraction kit, QIAprep spin mini prep; Qiagen Inc., Valencia, CA.
d.
Integrated DNA Technologies Inc., Coralville, IA.
e.
New England Biolabs, Ipswich, MA.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) declared that they received no financial support for the research, authorship, and/or publication of this article.