
Abstract
Mycoplasma hyorhinis has emerged as an important cause of systemic disease in nursery pigs. However, this bacterium can also be found in the upper respiratory tract of healthy swine. The current study describes the development of a quantitative polymerase chain reaction assay for the detection of M. hyorhinis and the evaluation of the assay in both disease diagnosis and disease surveillance using a large number of field samples. The analytical sensitivity was estimated to be 12 genome equivalents/μl. The assay was highly specific, detecting all 25 M. hyorhinis isolates tested and none of the 19 nontarget species tested. Assay repeatability was evaluated by testing different matrices spiked with known amounts of M. hyorhinis. Overall, assessment of the repeatability of the assay showed suitable precision within and between runs for all matrices. The coefficient of variation ranged from 10% to 24%. Mycoplasma hyorhinis DNA was detected in 48% of samples (pericardium, pleura, joints, nasal cavity, and lungs) from pigs with systemic disease. Mycoplasma hyorhinis was detected in nasal (92%) and oropharyngeal swabs (66%), as well as in oral fluids (100%). Potential uses of this tool involve the characterization of the prevalence of this pathogen in swine herds as well as bacterial quantification to evaluate intervention efficacy.
Introduction
Mycoplasma hyorhinis causes mainly fibrinous polyserositis and arthritis in pigs, and it has been implicated as a cause of pneumonia and otitis.2,21,25 This wall-less bacterium is a common finding in the upper respiratory tract of healthy pigs and is considered to be globally distributed.7,25,28 Stress and concomitant infection with other respiratory pathogens may predispose pigs to M. hyorhinis disease.10,13
Little is known about the epidemiology of this pathogen. The source of infection for young pigs is yet to be elucidated; however, it is presumed that sows can transmit the organism to their offspring, and subsequent transmission between pigs follows.15,22 To advance the knowledge of the dynamics of M. hyorhinis infection in affected herds, polymerase chain reaction (PCR) testing could be done on nasal swabs of pigs at the individual level or on oral fluids at the group level. However, because M. hyorhinis is a commensal of the upper respiratory tract of pigs, there is a need for a diagnostic tool that not only detects but also quantifies the amount of bacterial DNA in the sample.
Several diagnostic methods have been used for the detection of M. hyorhinis. Isolation of this pathogen requires special media, and the organism can take up to 14 days to grow.9,17 Immunohistochemical staining and fluorescent antibody tests have been used for detection of M. hyorhinis in tissues.11,16,19 Antibody-targeted assays have also been described in the literature.3,8,23 However, such assays have been employed for research purposes, they are not commercially available, and some require euthanasia of the animal. A number of PCR protocols targeting M. hyorhinis have been described.4,12,18 However, most of the protocols are based on traditional endpoint PCR protocols and therefore are not capable of quantifying the amount of bacterial DNA in a sample. Furthermore, the performance of these PCR protocols has not been evaluated with a wide range of clinical samples, and some of them were developed for the detection of M. hyorhinis in cell cultures only. 30
The current study describes the development and evaluation of a highly sensitive and specific quantitative PCR (qPCR) assay for the detection and quantification of M. hyorhinis in field samples. The use of this quantitative assay in epidemiological investigations will increase understanding of the dynamics of M. hyorhinis infection in swine populations.
Materials and methods
Mycoplasma hyorhinis isolates and culture conditions
A reference strain, M. hyorhinis ATCC 17981D, a was used for the development and standardization of the qPCR assay. In addition, 25 M. hyorhinis isolates cultured from clinical cases of polyserositis were tested. These isolates were recovered from different serosal surfaces such as pleura, pericardium, peritoneum, and synovial membrane. All M. hyorhinis isolates were cultured in modified Hayflick media incubated aerobically at 37°C for 2–14 days in 3-ml broth tubes and with 5% CO2 at 37°C on solid agar media. 17 Isolates were stored at −80°C following confirmation of growth on solid media.
Mycoplasma hyorhinis DNA isolation
Genomic DNA from all samples was obtained using a commercial kit according to manufacturer’s specifications. b The starting material for bacterial isolates was the growth from 4 plates harvested from solid media and resuspended in 0.5 ml of phosphate buffered saline (PBS) c or 0.5 ml of modified Hayflick media. The efficiency of the DNA extraction was assumed to be 100%. The final product was stored at −80°C and used as the DNA template.
Polymerase chain reaction primers and probe
The partial sequence of the 16S small subunit ribosomal (r)RNA gene of M. hyorhinis (GenBank accession no. GU227402.1) was aligned to other 16S rRNA partial sequences of closely related bacterial pathogens, including Mycoplasma hyopneumoniae (JN935889, GU227407.1, Y00149.1), Mycoplasma flocculare (GU227403), and Mycoplasma hyosynoviae (AY973563.1) using ClustalW 5 in MEGA. 29 Mycoplasma hyorhinis species–specific regions were selected to design forward and reverse primers, as well as the probe. Selected sequences were evaluated using Primer 3 software. d The fluorogenic probe was labeled with a fluorescent dye, 5-carboxyfluorescein (FAM) as reporter at the 5′-end and N,N,N′,N′-tetramethyl-6-carboxyrhodamine (TAMRA) dye as quencher at the 3′-end. e The ideal annealing temperature was 54°C.
Quantitative PCR conditions
The qPCR reaction for M. hyorhinis was prepared in a volume of 25 µl consisting of 5 µl of the template DNA, 12.5 µl of master mix solution, f 1 µl of reference dye (ROX), 0.125 µl of each 40 µM forward (MHF-5′-GATGTAGCAATACATTCAGTAGC-3′) and reverse (MHR-5′-AAGTGAAGCTGTGAAGCTC-3′) primers, 1.25 µl of 5 µM fluorogenic probe (5′-FAM-CGGATATAGTTATTTATCGCA-TAMRA-3′), and 5.0 µl of DNase and RNase-free water. The reactions were carried out on a real-time PCR system. g The cycling protocol consisted of 95°C for 3 min, followed by 35 cycles of 95°C for 15 sec and 54°C for 50 sec. All samples were run in duplicates, and water was used as the no target control in all reactions. The negative extraction control was 0.5 ml of PBS. c Absolute quantification of M. hyorhinis DNA was estimated from the extrapolation of the values from a 10-fold dilution standard curve included in each run.
Analytical sensitivity
Genomic DNA was extracted from M. hyorhinis ATCC 17981D, and the amount of DNA was quantified by spectrophotometry. h The number of genome equivalents (geq) was determined based on the genome size of M. hyorhinis (839,615 bp, approximately equivalent to 1 fg DNA per cell). With the objective of estimating the minimum number of geq that can be detected with the qPCR, 10- and 2-fold dilutions were made on nuclease-free water to achieve template concentrations from 107 to 100 geq/μl of extracted DNA. Results were compared to those obtained by a conventional endpoint PCR. 30
Analytical specificity
The basic local alignment search tool (BLASTn, http://www.ncbi.nlm.nih.gov/blast/Blast.cgi) was used to evaluate sequence similarity of the primers and probe to other targets. Nonspecific binding was evaluated by running the newly developed protocol by endpoint PCR. The presence of potential products resulting from nonspecific binding was assessed by electrophoresis on 1% agarose gels stained with ethidium bromide. In addition, a total of 19 reference strains of bacterial species commonly found in swine were tested with the qPCR: Actinobacillus equuli (NCTC 8529), Actinobacillus indolicus (NCTC 46KC2), Actinobacillus minor (NCTC NM305), Actinobacillus pleuropneumoniae (NCTC 4074), Actinobacillus porcinus (NM319), Actinobacillus rossii (NCTC 192), Bacillus cereus (NCTC 2599), Bordetella bronchiseptica (NRRL B-140), Erysipelothrix rhusiopathiae (NCTC 8163), Escherichia coli (laboratory strain), Haemophilus parasuis (NCTC 4557), M. flocculare (NCTC 10143), M. hyopneumoniae (NCTC 10110), M. hyosynoviae (NCTC S16), Pasteurella multocida (NCTC 10322), Salmonella enterica subsp. enterica serovar Choleraesuis (ETS 34), Staphylococcus aureus (Wichita), Streptococcus suis (958242), and Trueperella pyogenes (NCTC 5224). All strains were obtained from the American Type Culture Collection (ATCC), a with the exception of the A. indolicus, A. minor, A. porcinus, and E. coli strains, which belong to the strain collection of the Veterinary Diagnostic Laboratory of the University of Minnesota (VDL-UMN; St. Paul, Minnesota).
Repeatability (within-laboratory precision)
Evaluation of assay repeatability was carried out following the guidelines established by the Clinical and Laboratory Standards Institute (CLSI). 6 Spiked pericardium, nasal, oral fluid, and modified Hayflick medium (control) matrices were used to estimate the repeatability of the assay. A total of 20 nasal and pericardial swabs, as well as 10 ml of oral fluids were collected from designated pathogen–free (DPF) pigs. Swabs were pooled with 10 ml of PBS. c All matrices were M. hyorhinis negative by qPCR and endpoint PCR prior to the spiking. Matrices were spiked with a concentrated M. hyorhinis culture to achieve a high and a low bacterial concentration (106 and 103 geq/μl of extracted DNA, respectively). All samples used in the validation of the assay were extracted twice—at the beginning of the experiment and 5 days later following the protocol described above. Samples from each set of extractions were tested for 5 days. After the first 5 days, samples were discarded, and the second batch was extracted and tested for 5 days. Samples were run in triplicate, twice per day at different times. Each run employed a negative and positive extraction control as well as a PCR negative control (water).
Assay repeatability was evaluated by performing graphical analysis, estimating coefficient of variation (CV) values, and by using mixed-effects models to assess sources of variation. The CV values were estimated by using the equations reported in the CLSI guidelines for “Device or Laboratory Precision,” with the modification to increase the number of replicates from 2 to 3. 6 Data was log transformed, and mixed-effects models were used to estimate the mean response and variability associated with each dose by matrix combination. To explore how variances differed between combinations, models were fit to the cumulative data and then to each dose–matrix and dose–matrix–extraction combinations. The full model included concentration, matrix, extraction, and all interactions as main effects, with random effects for day, run, and triplicate. The models for each dose–matrix combination had extraction as main effect and day and run as random effects; the models for dose–matrix–extraction had day and run as random effects. All calculations were performed using R 3.0.2, lme4 package. i
Field samples
To compare the diagnostic performance of the qPCR with a previously published endpoint PCR, 30 a total of 49 clinical samples from pleura (14), peritoneum (3), pericardium (8), joint (18), nasal cavity (3), and lung (3) submitted to the VDL-UMN were tested. These samples originated from cases suggestive of M. hyorhinis–associated disease, evidencing fever, dyspnea, and reluctance to move. Discordant results between the 2 PCR protocols were fully investigated by reviewing pathological findings, and by sequencing the PCR product.
As well, 2 additional sets of samples from clinically healthy pigs were examined by qPCR alone. First, a total of 70 nasal swabs, 30 oropharyngeal swabs, and 34 oral fluid samples were taken from 8-week-old pigs. The pigs originated from herds with history of M. hyorhinis disease based on culture or detection by PCR; however, the pigs were clinically healthy at the time of sampling. Second, with the goal of testing a negative population, 24 pericardium and 10 nasal swabs were tested for M. hyorhinis by qPCR. These samples were obtained from adult pigs originating from a high biosecurity, DPF herd.
All samples were extracted following the procedure described above, with the exception of the starting material, which differed. For postmortem samples, the starting material was 200 μl of a solution resulting from vortexing a sample of fibrin from the affected serosal surface resuspended in 3 ml of PBS. c For samples from live pigs (nasal and oropharyngeal swabs) and from DPF pigs (pericardial and nasal swabs), swabs were suspended into 0.5 ml of PBS, c followed by vortexing and centrifugation. The remaining pellet was used for DNA extraction.
Sequencing
A subset of PCR products obtained from testing clinical samples and lower dilutions from the analytic sensitivity assay were purified using a commercial kit. j Samples were sequenced in both directions with the primers described above to obtain double coverage using a 96 capillary sequencer. k Obtained sequences were entered in BLAST for target confirmation.
Results
Analytical sensitivity and specificity
The analytical sensitivity of the M. hyorhinis qPCR was determined with a series of 10- and 2-fold dilutions of M. hyorhinis ATCC 17981D. The standard curve of M. hyorhinis DNA displayed linearity over 7 log units, as exemplified by a R2 > 0.99 for 10 consecutive setups. The undiluted initial DNA concentration measured by spectrophotometry was 23 ng/μl, which is equivalent to approximately 2.3 × 107 geq/μl. The detection limit was determined to be 12 geq/μl of extracted DNA (4.8 × 103 geq/ml of culture broth), which corresponded to a threshold cycle value of 34.2. The estimated qPCR efficiency was 91.29%. The length of the qPCR product estimated using an ethidium bromide–stained agarose gel was consistent with the expected length of 151 bp (data not shown). The PCR products from positive samples (lower dilutions) were sequenced. These sequences were analyzed by basic alignment using BLAST and were homologous to the M. hyorhinis 16S rRNA gene. The detection limit of the conventional endpoint PCR was the same as the qPCR.
Introduction of the primer and probe sequences in BLASTn did not reveal highly similar sequences other than M. hyorhinis. There was no evidence of nonspecific binding in a 1% ethidium bromide–stained agarose gel. The qPCR was highly specific, detecting exclusively M. hyorhinis isolates. All 19 non–M. hyorhinis bacterial isolates tested negative. In contrast, all 25 M. hyorhinis isolates from clinical cases tested positive.
Repeatability (within-laboratory precision)
The repeatability within and between runs and days appeared to be high and stable across all matrices (Fig. 1). There was a noticeable effect of the extraction on the estimated bacterial concentration, as evidenced by differences between the results obtained during the first 5 days (first extraction) compared with the last 5 days (second extraction); compared to the other matrices, oral fluids showed lower precision overall (Fig. 1). Considering the differences observed between the first and second extraction, CV estimates were calculated independently for each combination of matrix, dose, and extraction. The CVs ranged from 10% to 13% in the high concentration sample with the exception of the oral fluid matrix, extraction 1, which had a CV of 24%. The CVs ranged from 13% to 21% for the low concentration sample. Mixed models revealed 3 sources of variation, namely, “day,” “run-within-day,” and “replicate” (residual). Within the low-dose, “run” and “replicate” accounted for most of the variation, whereas in the high-dose, all 3 sources contributed to the observed variation.
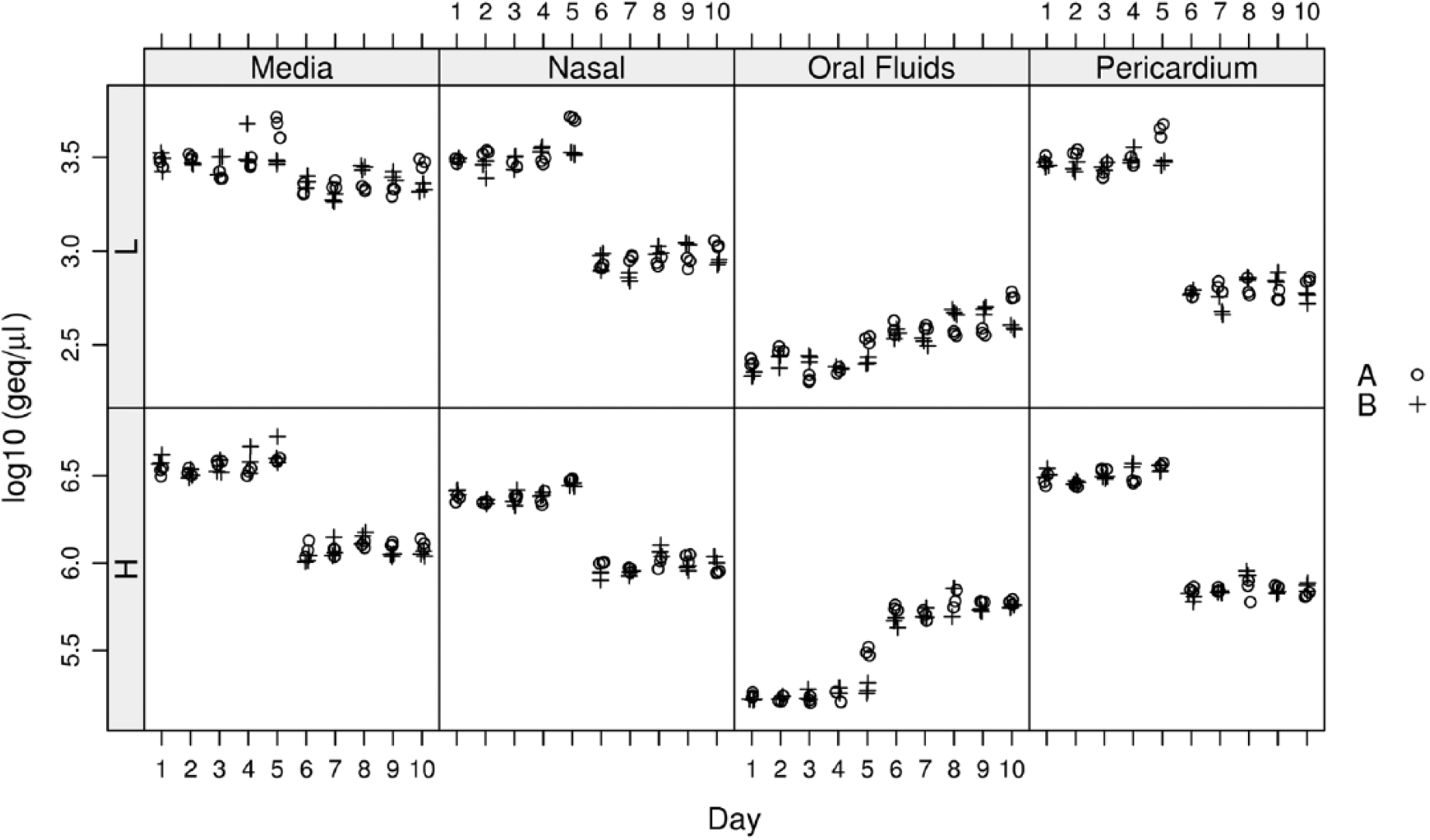
Panel of plots showing the bacterial load, expressed as genome equivalents per microliter, measured by the quantitative polymerase chain reaction assay, of 4 matrices spiked with 2 different Mycoplasma hyorhinis bacterial concentrations. Two extractions were performed. Samples from each set of extractions were tested for 5 days. Two plots are provided for each matrix based on 2 concentration levels (H = high level; L = low level). Circles (A) represent the replicates for the first run and crosses (B) represent replicates for the second run.
Field samples
The qPCR detected M. hyorhinis in 49% (24/49) of the samples from clinically affected pigs, compared to 35% (17/49) using the conventional endpoint gel-based PCR. The 7 samples with discordant results tested positive by qPCR and negative by gel-based PCR. Samples with discordant results had a low bacterial load (average 102 geq/μl) compared to the rest of the positive samples (average 104 geq/μl) and originated from multiple sampling sites (pleura, pericardium, joint, and nasal cavity). The PCR products of these samples were sequenced, and reliable DNA sequences were obtained for 6 of them. Sequence analysis of the discordant results confirmed the samples as M. hyorhinis, with 100% similarity.
Ninety-two percent (65/70) of nasal swabs, 66% (20/30) of oropharyngeal swabs, and 100% (34/34) of oral fluid samples of pigs originating from clinically affected herds tested positive for M. hyorhinis by qPCR. The average bacterial concentration of these samples was 105 geq/μl. All 24 pericardial swabs and 10 nasal swabs originating from adult DPF pigs were negative for M. hyorhinis.
Discussion
The goal of the current study was to develop a highly sensitive and specific qPCR for M. hyorhinis and to evaluate its application for diagnosis and surveillance with a wide range of sample types. The assay was shown to be highly sensitive, detecting 12 geq/μl of extracted DNA. This analytical sensitivity was comparable to that of a previously described endpoint PCR. The specificity of the assay was demonstrated by the lack of positives in a large panel of swine bacteria and by negative results obtained from clinical samples expected to be negative.
Assay repeatability was evaluated by testing different matrices spiked with known amounts of M. hyorhinis DNA. Different types of matrices were used to mimic diagnostic samples commonly tested in a diagnostic setting. Overall, assessment of the repeatability of the assay showed suitable precision within and between runs. Mixed models revealed various sources of variation (day, run, and replicate) with the majority of the variation due to run-within-day. The estimated coefficient of variation ranged from 10% to 24%. Similar values have been obtained with quantitative PCR assays targeting Mycoplasma genitalium and Mycoplasma haemocanis.1,27 There was an impact of less than 1 log of the extraction batch on the estimated amount of M. hyorhinis detected. This was an unexpected finding as sample extraction was performed both times following the exact same protocol. The current study, however, was not designed to evaluate the effect of extraction on qPCR results. The impact of extraction on assay reproducibility warrants further study. A slight decrease in the expected bacterial load observed with the oral fluids matrix might be due to overall decreased extraction efficiency due to the sample composition or to the presence of PCR inhibitors.
Previously published endpoint PCR assays were developed for the detection of M. hyorhinis in cell culture, 30 in lung samples,14,26 or in formalin-fixed, paraffin-embedded tissues from pigs with polyserositis. 12 In a 2014 published study, a qPCR was developed with a reported detection limit of 125 geq/μl. 31 However, the use of the assay was only assessed using bacterial isolates and not clinical samples, and assay repeatability was not evaluated. 31 In the current study, M. hyorhinis DNA was detected in multiple sample types originating from clinically (pleura, pericardium, peritoneum, joint, nasal, and bronchial swabs) and nonclinically affected pigs (oral fluids, oropharyngeal, and nasal swabs). The proportion of positive samples from pigs with disease (pleura, pericardium, peritoneum, nasal cavity, and lung) observed in the current study (48%) is in accordance with that observed in other reports. In one previous study, M. hyorhinis was isolated from 35% of serosal surfaces of pigs submitted to a diagnostic laboratory. 8 Approximately 55% of serosal tissues from affected pigs submitted to the VDL-UMN were positive by endpoint PCR. 24
The high proportion of qPCR-positive oral fluids, and nasal and oropharyngeal swabs from clinically healthy pigs was expected because M. hyorhinis is a commensal organism of the upper respiratory tract of pigs. To the authors’ knowledge, the detection of M. hyorhinis in oral fluids has not been previously assessed. While an in-depth validation of this sampling technique for detection of M. hyorhinis is currently needed, the practicality and noninvasiveness nature combined with the ability to perform herd level surveillance, makes this technique promising. 20 The lack of detection of M. hyorhinis in pericardial and nasal swabs from DPF pigs was anticipated because these were clinically healthy adult pigs with no lesions at necropsy and originating from negative herds. In addition to the origin of the DPF pigs, the nondetection of M. hyorhinis in nasal swabs could be due to as-yet-unidentified host factors. For example, it is possible that older pigs are able to clear M. hyorhinis infection more efficiently than younger pigs.
In summary, a rapid, high-throughput, sensitive, and specific assay for detection of M. hyorhinis in multiple sample types is described. The results of the study demonstrate the potential of the assay for accurate detection of M. hyorhinis in field samples. Furthermore, the assay shows a potential for use in characterizing the nasal colonization dynamics of this pathogen in different ages groups, to evaluate bacterial shedding over time, and to evaluate the efficacy of treatment and prevention protocols.
Footnotes
Acknowledgements
The authors thank Dr. Yin Jiang (University of Minnesota, Veterinary Diagnostic Laboratory) for her technical assistance.
a.
Strain 17981D, American Type Culture Collection, Manassas, VA.
b.
DNeasy blood & tissue kit, Qiagen Inc., Germantown, MD.
c.
Sigma-Aldrich, St. Louis, MO.
e.
TaqMan, Life Technologies, Grand Island, NY.
f.
QuantiFast Probe PCR kit, Qiagen Inc., Germantown, MD.
g.
ABI 7500 fast, Life Technologies, Grand Island, NY.
h.
NanoDrop 2000, NanoDrop, Wilmington, DE.
j.
QIAquick PCR purification kit, Qiagen Inc., Germantown, MD.
k.
ABI 3730xl BigDye Terminator v 3.1, Life Technologies, Grand Island, NY.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.