
Abstract
The application of the original Koch postulates and the molecular Koch postulates in the definition of the etiological agents of polymicrobial diseases has received little or no attention. In the present study, denaturing gradient gel electrophoresis (DGGE) of oral samples (n = 3) from each of 3 categories of animals (healthy, diseased [gingivitis], and then oxytetracycline-treated) was used and revealed different bacterial community structures in a model polymicrobial disease (gingivitis) and after clinical cure. Potential microbes associated with the disease and belonging to the following families were identified: Fusobacteriaceae, Porphyromonadaceae, Flavobacteriaceae, Alcanivoracaceae, Bacteroidaceae, Xanthomonadaceae, and Neisseriaceae. Liquid chromatography–mass spectrophotometric analysis of culturable anaerobic bacteria culture supernatant revealed 3 major compounds (2-hydroxycaproic acid, phenyllactic acid, and indole acetic acid) that differentiated the healthy and disease groups. Results indicate that different microbial community structures were associated with the healthy and disease oral states. The results demonstrate the potential of DGGE as a tool in the detection and designation of etiological agents of polymicrobial diseases.
Keywords
Polymicrobial diseases are diseases associated with multiple etiological agents that act synergistically or in succession in the pathogenesis of the disease and are often referred to by physicians and veterinarians as mixed or complex infection. 2 The identification of the pathogens involved in a polymicrobial disease is necessary for the accurate management of such diseases.
According to the Koch postulates, an etiological agent should be present and able to cause a specific disease; the agent should be able to cause disease when introduced in a healthy individual and to be isolated from the same individual in a pure culture. 4 These postulates have been readily adapted to diseases associated with a single and specific etiological agent. However, little or no attention has been given to disease conditions associated with polymicrobial etiology.
In recognition of the limitations of the original Koch postulates and the redefinition of the principles of etiology based on the molecular Koch postulates, 4 assertions (congruence, consistency, cumulative dissonance, and curtailment) for the “process of etiological certainty” have been proposed. 7 The assertions generally redefined the criteria for the association of an etiological agent with a disease based on modern tools that were not available at the time the original Koch postulates were made.
Gingivitis is a model polymicrobial disease and has been suggested to be a result of microbial ecological flux in the oral cavity. 3 In the present study, based on the prescribed assertions relating to “process of etiological certainty,” 7 cases of gingivitis in macropods were evaluated using antibiotic administration and denaturing gradient gel electrophoresis (DGGE) in order to establish the application of the molecular Koch postulate in the definition of the etiological agents in a polymicrobial disease situation. Liquid chromatography–mass spectrophotometry (LC-MS) was performed in order to evaluate the bacterial metabolite profiles of pooled samples from healthy and diseased macropods.
Oral swab samples were collected (in duplicate) from the gingival margin of the premolar and molar tooth of 3 randomly selected healthy yellow footed rock wallabies (YFRW; or ring-tailed rock wallabies; Petrogale xanthopus) and 3 YFRW with gingivitis during routine health checks at the Monarto Zoo, South Australia. The presence of gingivitis was established by the observation of swelling and redness of the gum, bleeding of the gum and signs of pain on swabbing, and the show of reluctance to feeding. The 3 wallabies with gingivitis were treated with 20 mg/kg of long-acting oxytetracycline a administered intramuscularly (6 times, at intervals of 3 days) as part of the routine health checks at the zoo. A second set of samples was collected from the same set of treated animals 3 weeks after antibiotic administration was completed. The samples were stored at −20°C for further analysis. All samples were analyzed within 3 days of storage. Approval for use of the samples was granted by the Animal Welfare Sub-Committee of the School of Biological Science, Flinders University of South Australia.
DNA was extracted from 1 swab from each animal using a commercial kit
b
according to the manufacturer’s instructions. Template DNA (1 μl [10 ng]; replaced with 1 μl of nuclease-free water for the negative control) was amplified in a total volume of 25 μl consisting of 12.5 μl of commercial mastermix
c
and an aliquot (1 μl) each of 10 μM forward primer (with GC-Clamp [underlined] to enhance DGGE band separation): 341FGC: 5’-
The polymerase chain reaction (PCR) cycle conditions consisted of an initial denaturation of 95°C for 5 min, followed by 10 cycles of 94°C for 30 sec, 65°C for 30 sec, 72°C for 1 min; 10 cycles of 94°C for 30 sec, 60°C for 30 sec, 72°C for 1 min; 10 cycles of 94°C for 30 sec, 55°C for 30 sec, 72°C for 1 min, and a final extension at 72°C for 10 min. To assess PCR specificity, electrophoresis of the PCR products was carried out in 2% agarose gel in 0.5× Tris–acetate–EDTA (TAE; pH 8) buffer and run at 80 V for 60 min and stained with a commercial nucleic acid dye d according to the manufacturer’s instructions.
The PCR products (15 µl) were fractionated (in triplicate) by DGGE e on 6% polyacrylamide f with a 45–60% formamide gradient. Electrophoresis was conducted at 60 V for 18 hr at 60°C in a 1× TAE buffer. Five microliters of 1-kb molecular weight marker was also loaded into the gel and used as reference ladder to compare the position of each band between individual samples. All samples (healthy, diseased, and treated) were analyzed on the same gel. The DGGE gel was then stained with silver nitrate as previously described. 6 As Fusobacterium necrophorum is thought to be one of the major etiological agents of periodontal disease in macropods,11,12 the presence of this organism was investigated in all categories of samples by cycliplex PCR as described previously. 1
Analysis of the stained DGGE gel was carried out by digital scanning, g and an evaluation of the oral microbial community structure associated with healthy, disease, and treatment categories was performed using image analysis software. h An unweighted pair group method with mathematical averages (UPGMA) algorithm was used to generate dendrograms, which were then used for the assessment of similarities between microbial communities in each group of animals evaluated.
Dominant bands in the DGGE profile representing operational taxonomic units (OTUs) of bacteria associated with specific conditions were identified by both digital software h and by visual inspection (with the aid of white light) of the silver-stained polyacrylamide gel. The identified OTUs were then excised from the polyacrylamide gel using a single sterile scalpel per band. Excised bands were washed 3 times in sterile water by centrifugation at 14,000 × g for 5 min, and DNA was eluted by overnight incubation at 60°C in 50 μl of fresh sterile, DNA-quality water. The eluted DNA was reamplified with 341F (without the GC-Clamp) and 518R primer sets in a single PCR reaction as described previously; electrophoresis of the PCR product was carried out to confirm reaction specificity as described previously. The reamplified PCR product was then cloned into a vector using a commercial cloning kit, i according to the manufacturer’s instructions. The transformation mix (100 μl) was spread on nutrient agar plates containing 100 mg of ampicillin for selection of positive clones and then incubated at 37°C for 24 hr.
The resulting PCR products were purified j using a commercial kit according to the manufacturer’s instructions and submitted to a commercial service provider for sequencing. k Analysis of the oligonucleotide sequences was carried out using BLASTn algorithm at the National Centre for Biotechnology Information (NCBI) website (http://blast.ncbi.nlm.nih.gov/).
In order to verify that the clones obtained from the excised DGGE bands were correct, a sterile 10-µl pipette tip was used to stab the midsection of each band of interest (from the replicate) in the original DGGE gel. This was then used as a DNA template in another PCR assay (without the GC-clamp) as described previously. The PCR products were purified and sequenced directly, and the sequences were matched with that of the corresponding cloned excised bands by using the BLASTN algorithm. Matched sequences suggested that the DGGE bands occurring at the same position in the gel had similar sequence information.
The second oral swab from the 3 randomly selected healthy wallabies and the 3 wallabies showing clinical signs of gingivitis were pooled according to the individual categories (healthy and disease categories). The pooled samples from each category were inoculated into 40 ml of broth medium (pH 6.8) and incubated in an anaerobic jar containing anaerobic maintenance agent l at 37°C for 48 hr. The broth was composed of brain–heart infusion m (37 g/l), yeast extract n (5 g/l), tryptone o (10 g/l), and sodium thioglycolate (1 g/l). p
The concentration of bacterial cells in the 48 hr–incubated broth culture was adjusted to McFarland standard no. 3, q and 20 μl was then inoculated into 70 ml of broth medium in a 100-ml (1 per category) bottle, which was then sealed with a rubber stopper and metal cap to prevent gas escape. The broth culture was then incubated anaerobically at 37°C for 72 hr. The broth culture was then centrifuged r at 1,902 × g at 4°C for 30 min to pellet the bacterial cells. The supernatant was removed, filtered through a 0.2-μm filter, s and stored at 4°C for LC-MS analysis. All manipulations were carried out in an anaerobic chamber purged with 70% nitrogen and 30% carbon dioxide.
The LC-MS analysis was performed with a mass spectrophotometer t consisting of a 2695 separation module and 2487 dual wavelength ultraviolet (UV) light detector; the liquid chromatographic u separation was achieved using a commercial column v with the dimensions of 2 mm ID × 150 mm length. Twenty microliters of the cell-free culture supernatants were analyzed at a flow rate of 0.2 μl per min in a mobile phase consisting of 0.5% aqueous formic acid and acetonitrile. The gradient (%) consisted of t = 0 min 95% A, t = 1 min 95% A, t = 18 min 0% A, t = 23 min 0% A, t = 25 min 95% A, and t = 30 min 95% A. The UV detection wavelengths were 230 and 280 nm.
Four MS scan functions were employed, 2 scans in the positive ion mode at low (35 V) and high (70 V) sample cone voltages and 2 in the negative ion mode at low (30 V) and high (70 V) sample cone voltages. Data were collected in electrospray MS mode using a commercial data system for both the MS and UV data. The LC-MS analysis was repeated to assess reproducibility by analyzing a fresh cell-free culture supernatant from the same set of samples. Tentative identification of the compounds of interest was achieved by a search of the LC-MS–identified peak characteristics against databases such as METLIN, KEGG, HMDB, and LIPIDMAPS.
Clinical observation of the oxytetracycline-treated animals showed resolution of the clinical signs and symptoms of gingivitis after 3 weeks post antibiotic administration. The swelling, bleeding, and signs of pain were not observed at this time, and the animals were apparently healthy. From the DGGE gel (Supplemental figure), a UPGMA-generated dendrogram was produced (Fig. 1), which revealed a 40% similarity coefficient between the healthy category and each of the disease and treated categories. The similarity coefficient between the disease and treated categories was 53%.
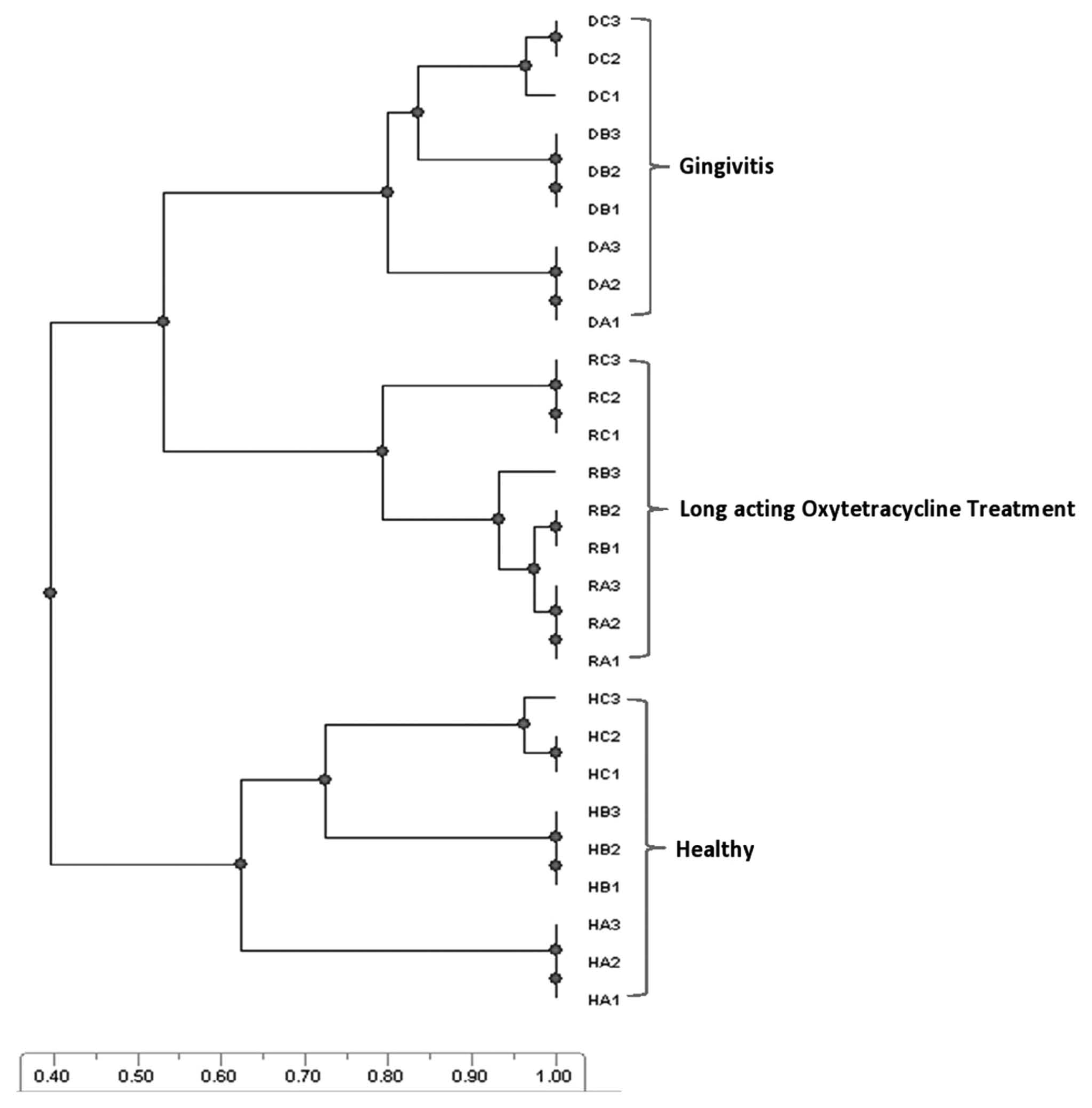
The unweighted pair group method with arithmetic mean (UPGMA) dendrogram of denaturing gradient gel electrophoresis fingerprints of healthy, disease (gingivitis), and long-acting oxytetracycline–treated categories. The healthy category shares 40% similarity coefficient with each of the disease and oxytetracycline-treated categories, which both share a 53% similarity coefficient. Each sample (n = 3) was fractionated in triplicate (HA1–HA3 = triplicate of the same sample; HB1–HB3 = triplicate of the second sample; HC1–HC3 = triplicate of the third sample). H = healthy; R = antibiotic treatment; D = gingivitis.
Table 1 shows bacterial community members that were dominant and associated with the gingivitis category but not with the healthy and long-acting oxytetracycline treatment categories. By applying the molecular Koch postulate, which defines an etiological agent by “curtailment of infective process on controlled introduction of biomedical intervention,” 7 the polymicrobial species associated with gingivitis in the assessed animal subjects were defined to include: F. necrophorum (Fusobacteriaceae), Porphyromonas gulae (Porphyromonadaceae), Chryseobacterium sp. (Flavobacteriaceae), Alcanivorax sp. (Alcanivoracaceae), Bacteroides pyogenes (Bacteroidaceae), Stenotrophomonas maltophilia (Xanthomonadaceae), and Neisseria zoodegmatis (Neisseriaceae; Table 1). The nucleotide sequences were deposited in the NCBI database with GenBank accession nos. JX891666–JX891672. The bands (OTUs) representing these bacterial species were not observed in the healthy group or the diseased group after administration of the long-acting oxytetracycline. Fusobacterium necrophorum DNA was detected by PCR only in the gingivitis-associated samples and was not detected in the healthy or treated animals, suggesting that F. necrophorum and possibly other DGGE-identified microbes in the disease categories are associated with gingivitis.
Dominant bands (bacterial species) identified from denaturing gradient gel electrophoresis clone library of the gingivitis category. The fingerprints of these clones were not observed after oxytetracycline treatment and in the healthy category.*

Information in parentheses is the GenBank accession number.
The metabolite profiles of the combined bacterial community metabolism, including the positive and negative ions distributions in the healthy and periodontal disease categories, were different (Fig. 2). Most importantly, the LC-MS profiling revealed peaks of compounds (Cpd1, Cpd2, and Cpd3) that differentiated the bacterial communities in the healthy and disease categories. These compounds were more abundant and distinct compared to other peaks in the disease category (Fig. 2B) and had nominal masses (mass-to-charge ratio [m/z]) and abundances (%) of 131 (60%), 147 (80%), and 132 (90%), respectively, with high-performance liquid chromatography retention times recorded as 12.12 min, 13.23 min, and 14.49 min, respectively. The peaks were tentatively identified as 2-hydroxycaproic acid (Cpd1), phenyllactic acid (Cpd2), and indole acetic acid (Cpd3). The peaks did not appear in the healthy category (Fig. 2C). The results observed were reproducible.
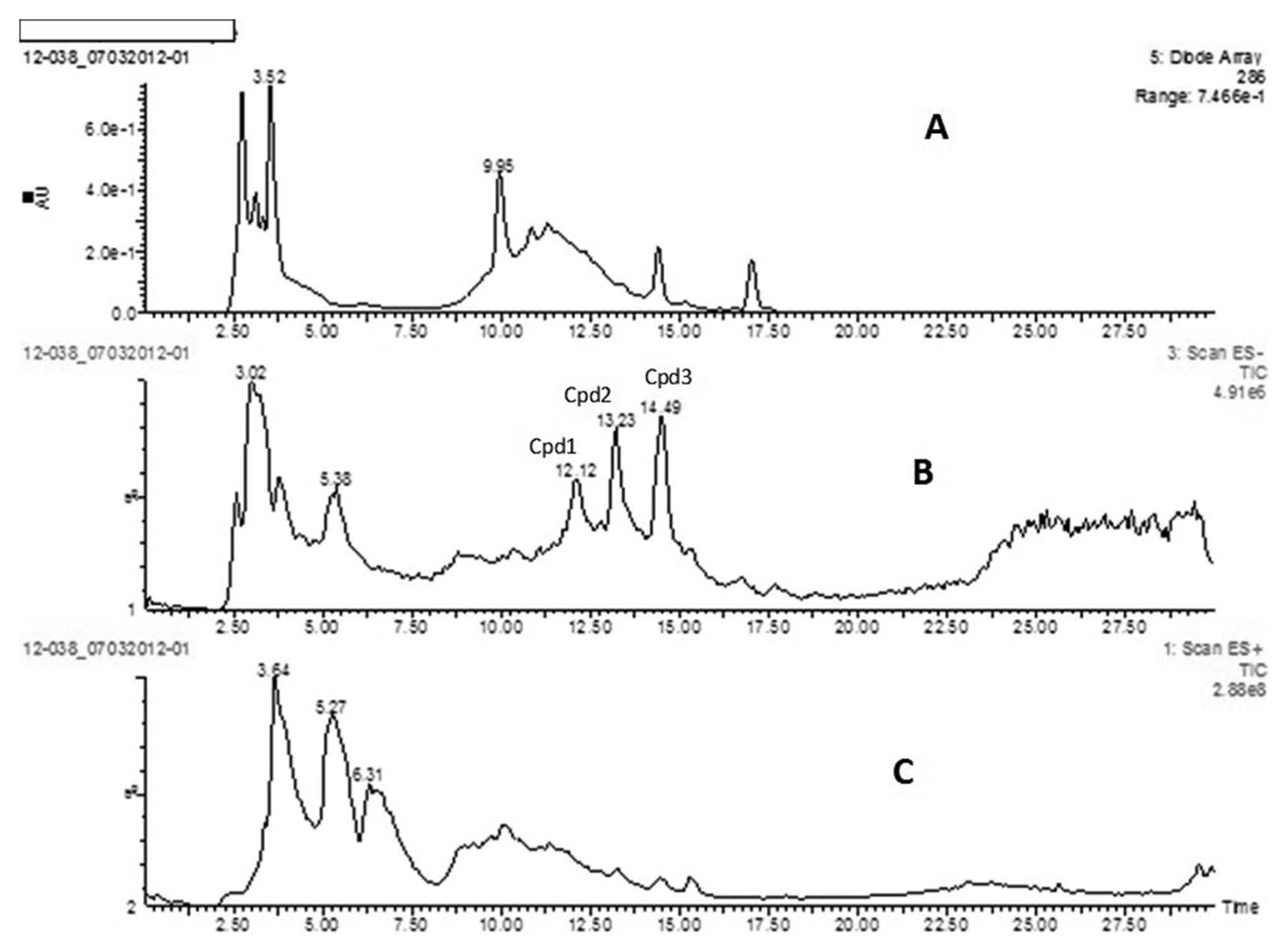
Liquid chromatography–mass spectrophotometry traces of anaerobic bacteria cell-free culture supernatants from pooled samples: A = control (without bacterial inoculums); B = anaerobic bacteria cell-free culture supernatants from disease group; C = anaerobic bacteria cell-free culture supernatants from healthy group. The peaks appearing at retention time 12.12 min (Cpd1 = 2-hydroxycaproic acid), 13.23 min (Cpd2 = phenyllactic acid), and 14.49 min (Cpd3 = indole acetic acid) on trace B represent the profile that differentiate the healthy from the disease group.
This study revealed that the bacterial communities as shown by a UPGMA-generated dendrogram of the DGGE fingerprint of the healthy, disease, and treated categories were different (see Supplemental figure). The LC-MS result suggested that the differences in the bacterial community structure as shown by the DGGE analysis may have accounted for the different metabolic profile observed. Moreover, this indicated dissimilar metabolic pathways for these different bacterial community structures, as the metabolites resulted from bacterial interactions at the community level. 9
The wallaby population investigated in the present study was part of a breeding program. Therefore, a control group of healthy wallabies to which long-acting oxytetracycline is administered could not be included in the study. A control group would have provided data on the effect of antibiotic treatment on the normal bacterial community structure in healthy subjects for comparison with the posttreatment gingivitis category. However, the important observation is that clinical cure of gingivitis after antibiotic treatment was associated with changes in bacterial community structure and that known periodontopathogens associated with gingivitis were not detected in the healthy and posttreatment gingivitis categories.
Members of the bacterial genus including Bacteroides, Fusobacterium, and Porphyromonas have been isolated from periodontal disease cases in previous studies.11,12 The previous studies were based on culture-dependent methods, which could not identify bacteria taxa that are unculturable and may require the use of stringent growth conditions in order to isolate fastidious microbes. However, the method reported in the current study could differentiate bacterial species (including fastidious bacteria) in healthy and diseased animals, in a single polyacrylamide gel, and fulfills the fourth assertion of the molecular Koch postulate. 7 The method also helped to circumvent the limitation of culture-dependent methods, which includes the inability to culture and identify slow-growing and/or fastidious organisms. 5 It is noteworthy that some of the microbes identified are members of the normal oral flora in human beings and animals and are opportunistic pathogens that increase in number, thereby becoming dominant in conditions that favor the establishment of periodontal disease. 8
The results presented in the current study demonstrate the application of the molecular Koch postulate in a polymicrobial disease situation and show that the DGGE method has the potential for elucidating polymicrobial disease etiology. However, the limited number of samples investigated reduced the power of the study and thus calls for a more comprehensive investigation.
Footnotes
Acknowledgements
The authors thank Dr. Ian Smith and the veterinary nurses in Adelaide and Monarto Zoos, South Australia for assistance with sample collection and advice.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by Flinders University of South Australia. John F. Antiabong was supported by a Flinders University IPRS scholarship.
a.
Long-acting oxytetracycline hydrochloride, Pfizer Australia Pty Ltd., West Ryde, New South Wales, Australia.
b.
Instagen matrix, Bio-Rad Laboratories Pty Ltd., Gladesville, New South Wales, Australia.
c.
Gotaq Green mastermix, Promega Australia, Alexandria, New South Wales, Australia.
d.
Invitrogen SYBR Gold, Life Technologies Australia Pty Ltd., Mulgrave, Victoria, Australia.
e.
Dcode Universal Mutation Detection System, Bio-Rad Laboratories Pty Ltd., Gladesville, New South Wales, Australia.
f.
Polyacrylamide gel, Bio-Rad Laboratories Pty Ltd., Gladesville, New South Wales, Australia.
g.
Epson 2700 scanner, Epson Australia Pty Ltd., Sydney, New South Wales, Australia.
h.
TotalLab TL120 software, Phoretix Ltd., Newcastle Upon Tyne, United Kingdom.
i.
TOPO TA Cloning Kit for Sequencing, Invitrogen Corp., Carlsbad, CA.
j.
Wizard SV gel and PCR clean-up system, Promega Australia, Alexandria, New South Wales, Australia.
k.
Australia Genome Research Facility, Adelaide, South Australia, Australia.
l.
Oxoid Anaerogen, Thermo Fisher Scientific Australia Pty Ltd., Adelaide, South Australia, Australia.
m.
Oxoid brain heart infusion, Thermo Fisher Scientific Australia Pty Ltd., Adelaide, South Australia, Australia.
n.
Oxoid yeast extract, Thermo Fisher Scientific Australia Pty Ltd., Adelaide, South Australia, Australia.
o.
Oxoid tryptone, Thermo Fisher Scientific Australia Pty Ltd., Adelaide, South Australia, Australia.
p.
Sodium thioglycollate, Sigma-Aldrich Pty Ltd., Sydney, New South Wales, Australia.
q.
Oxoid McFarland standard no. 3, Thermo Fisher Scientific Australia Pty Ltd., Adelaide, South Australia, Australia.
r.
Sigma 3-16PK refrigerated centrifuge, Sartorius Stedim Australia Pty Ltd., Melbourne, Victoria, Australia.
s.
0.2 μm filter, Millipore, Kilsyth, Victoria, Australia.
t.
Micromass Quattro micro, tandem quadrupole mass spectrometer, Waters, Manchester, United Kingdom.
u.
Waters liquid chromatograph, Waters Corp., Milford, MA.
v.
SGE Wakosil C18 column, SGE, Ringwood, Australia.
w.
Masslynx data system, Waters, New South Wales, Australia.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.