
Abstract
Cell line cross-contamination as well as genetic drift during passaging have been acknowledged as widespread problems since the 1960s. Improper cell line identification can invalidate results and, if not discovered, pollute the scientific community’s body of knowledge with regard to cancer cell lines, their gene expression, and their drug susceptibilities. Despite the obvious need, validation of cell line identity is not yet widely required, and the problem persists. A highly sensitive polymerase chain reaction (PCR)-based approach and short tandem repeat (STR) profiling were used to examine the prevalence of inter- and intraspecies cell line contamination in a veterinary research setting. First, 60 cell lines from 6 laboratories were tested with multiplex species-specific PCR capable of identifying 6 commonly used species. Of these, 3 were determined to be misidentified by species. Second, to identify intraspecies contamination among canine cancer cell lines, 29 canine lines from 3 different laboratories were analyzed with STR fingerprinting. Using this methodology, 3 canine cell lines were determined to be misidentified or cross-contaminated by other canine cell lines. Finally, genetic drift was observed within 1 cell line obtained from different laboratories. These findings emphasize the importance of cell line validation as a critical component of “good cell culture practice.” A database of the STR profiles obtained in the current study has been established for future comparison and validation of canine cell lines by investigators at Colorado State University and other institutions.
Cell lines are widely used in biomedical research as in vitro models for disease. HeLa cells were established in 1952 and, as early as 1958, interspecies cross-contamination between these cells and other lines was observed. 1 In the 1960s, karyotyping, examination of biochemical polymorphisms, and immunological approaches were developed to test cell line identity 6 within and between species but the early pioneers of cell line validation generally met with resistance from the scientific community. 1 More recently, it has been estimated that 18–36% of cell lines may be contaminated or misidentified. 9 With the advent of relatively simple polymerase chain reaction (PCR)-based DNA fingerprinting techniques, a new drive for cell line validation was initiated by Roland Nardone with the circulation and, later, publication of a white paper aimed at providing a solution for eradication of cell line misidentification. 11
In addition to misidentification, problems with excessive subculturing of cells have also been identified. 7 As cell lines are maintained in culture for long periods of time, selective pressures are being exerted on them and, thus, lines are subject to genetic drift, especially at higher passage numbers. Therefore, even when a cell line has not been contaminated, it can take on new attributes, skewing experimental data. Currently, an increasing number of journals require cell line validation prior to publication as examples emerge of cell lines with false identities being used and published long after they have been identified as problematic. 1
Diseases in veterinary patients are becoming widely acknowledged as valid translational models for similar diseases in human beings and, as such, veterinary research facilities are performing an increasing number of in vitro and in vivo clinical trial studies using models from a variety of species. Thus, it is crucial for the veterinary community to undertake the necessary steps to validate all cell lines in use and implement guidelines that facilitate future validation when creating new cell lines.
Researchers at the Colorado State University (CSU) Animal Cancer Center (ACC) use a variety of cell lines from a number of different species, including a panel of canine cancer cell lines. Because of this diversity, it was determined that testing for both inter- and intraspecies cell line contamination was necessary. Multiplex PCR has been demonstrated to be effective at determining species contamination, 4 and short tandem repeat (STR) profiling is commonly used in animal breed detection and parentage testing as well as human forensics to differentiate between individuals of the same species. Utilizing these relatively simple and robust methods, the current study has identified instances of both inter- and intraspecies cell line contamination. Sixty cell lines from multiple species of origin, 29 of which were canine, were tested via PCR. Subsequent STR screening of the 29 canine cell lines has allowed development of a database of canine STR profiles for comparison to other investigators’ cell lines and periodic cell line revalidation.
Considering the variety of species of cell lines currently in use at the ACC, a multiplex PCR test was adapted from previous work to differentiate among cell lines from dogs, cats, mice, rats, Chinese hamsters, and human beings. 4 Genomic DNA from 60 cell lines from 6 laboratories with multiple presumed species of origin was extracted per the manufacturer’s protocol a from cells grown to 90% confluency on 10-cm culture dishes or from pelleted cells. For the purposes of comparison, this screen tested cell line duplicates from different laboratories. The PCR reactions were performed using 1 µl (50–400 ng) of genomic DNA (gDNA) in a reaction mixture adapted from previous work 4 containing 20 mM Tris–HCl (pH 8.4), 50 mM KCl, 6 mM MgCl, 0.5% glycerol, 0.006% NP40/Tween 20 (1:1 v:v), 500 mM each deoxyribonucleotide triphosphate, primers, and 1.25 U polymerase. b Primers were commercially synthesized c ; sequences and concentrations were as previously published 4 with the exception of the Chinese hamster antisense primer, which was adapted to 5’-GCGTAGGCGAACAGGAAGTATC-3’ to match the currently published genomic sequence. Internal control primers that detect a 70-bp amplicon in all species were also included. Thermal cycling conditions were: 95°C for 5 min followed by 30 cycles of 95°C for 30 sec, 60°C for 15 sec, and 72°C for 30 sec, and completed with a 7-min final elongation at 72°C. The PCR products were run on a 2% agarose gel in Tris–borate–ethylenediamine tetra-acetic acid at 100 v for 60 min and visualized under ultraviolet light by ethidium bromide staining. A ladder containing PCR of gDNA from all 6 species was run concurrently. To validate the sensitivity of the multiplex PCR, each species’ DNA was artificially contaminated with 10% and 1% of another species’ DNA and tested.
Short tandem repeat profiling was performed on cell lines that PCR testing identified as canine in origin to assess the frequency of intraspecies contamination among cell lines. One µl of gDNA prepared as above was input into multiplex STR PCR reactions per the kit manufacturer’s protocol. d Two separate runs were performed to test a total of 29 cell lines (20 presumed unique lines, 9 duplicates from different laboratories). A positive control sample from the kit was included in each run. Thermal cycling conditions were as recommended by the manufacturer without controlled ramping. The PCR products were analyzed via capillary electrophoresis e per manufacturer’s protocols except as follows: 1 µl of PCR product was mixed with 9 µl of water to lower signal intensity during the run, and 1.5 µl of diluted PCR product was then mixed with 0.5 µl size standard f and 10 µl of highly deionized formamide. Additionally, POP7 polymer was used instead of POP4, and the array length was 50 cm. Run conditions were identical to the default run module except the injection time was increased from 15 to 24 sec, and the scan number was shortened from 1,800 scans to 1,750. Data interpretation was performed by manually binning alleles into size groupings and assigning allele designations.
The multiplex PCR was successfully able to identify all 6 tested species and the internal control amplicon at previously published amplicon sizes (Fig. 1A). 4 Furthermore, when each species of interest was contaminated with 10% and 1% gDNA from a different species, the PCR detected this contamination with 100% success rate. Figure 1B demonstrates this contamination study with canine gDNA as the primary input alone (lane 1) and contaminated with 10% (even lanes) and 1% (odd lanes) of human (lanes 2 and 3), cat (lanes 4 and 5), Chinese hamster (lanes 6 and 7), rat (lanes 8 and 9), and mouse (lanes 10 and 11) DNA. Lane 12 is a negative control with no input gDNA, and the final 2 lanes are the species ladder and a 100-bp ladder, respectively.
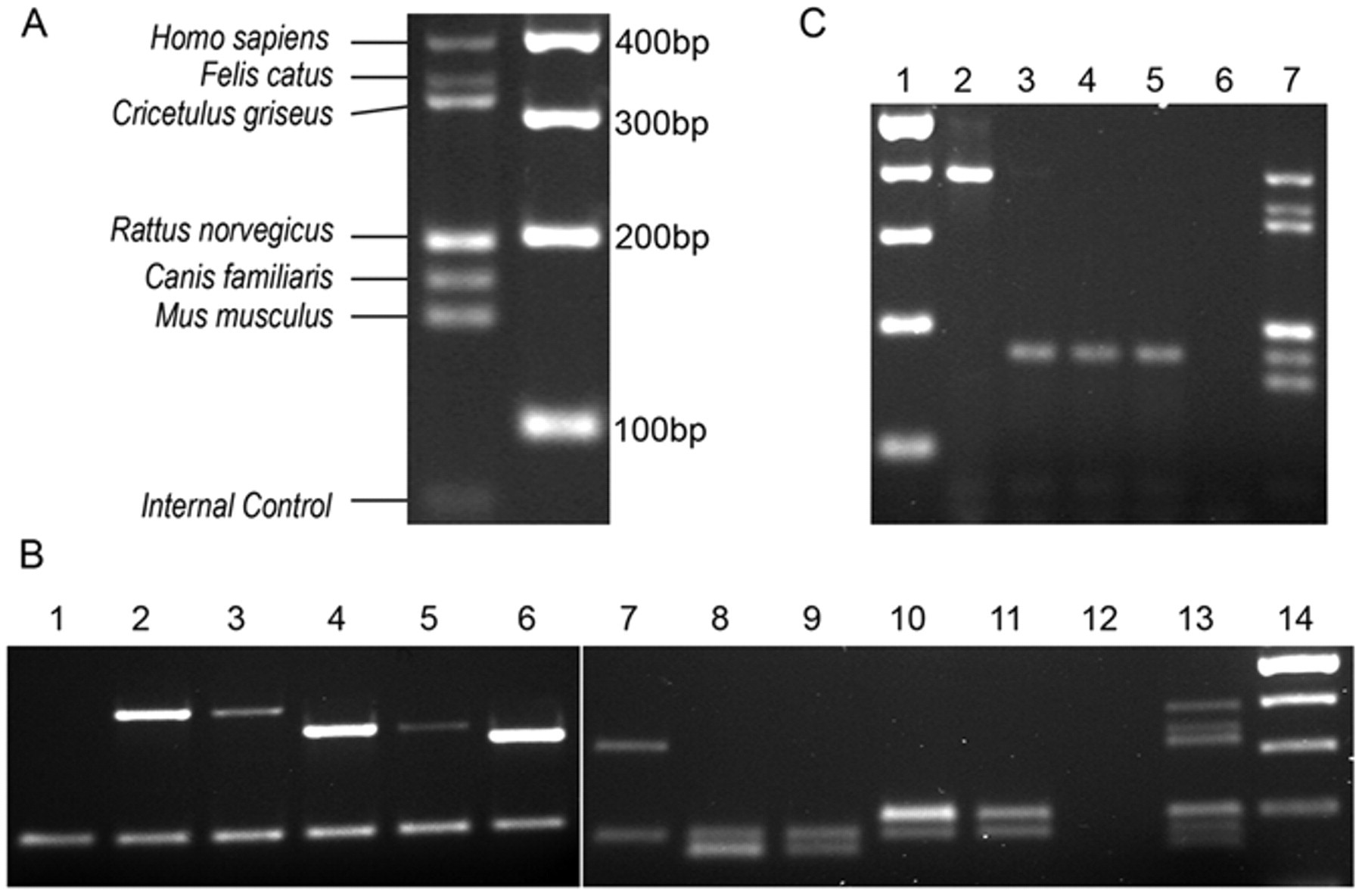
Multiplex polymerase chain reaction agarose gel electrophoresis.
Multiplex PCR testing of a total of 60 cell lines yielded 3 cases of mistaken identity. The first, a slow-growing canine osteosarcoma cell line “Yamada” was identified by PCR as murine in origin. A sample of this cell line from a different laboratory tested as canine indicating that the first laboratory had experienced a previous mislabeling or contamination event. The second case of mistaken identity was observed in another slow-growing canine cell line: the “Parks” melanoma tested as human in origin by PCR (Fig. 1C). The investigator working with this line had noticed a change in morphology and proliferation rate and, thus, requested the validation. As with the “Yamada” cell line, a different laboratory’s stock of the “Parks” cell line tested as canine. Finally, another presumed canine osteosarcoma line, “Grey,” was identified by PCR as human in origin. None of these 3 samples demonstrated any evidence that the original species was still present in the sample. This result was not surprising, as previous studies have shown that contaminating cell lines can completely overgrow cultures in as few as 4 or 5 passages. 1
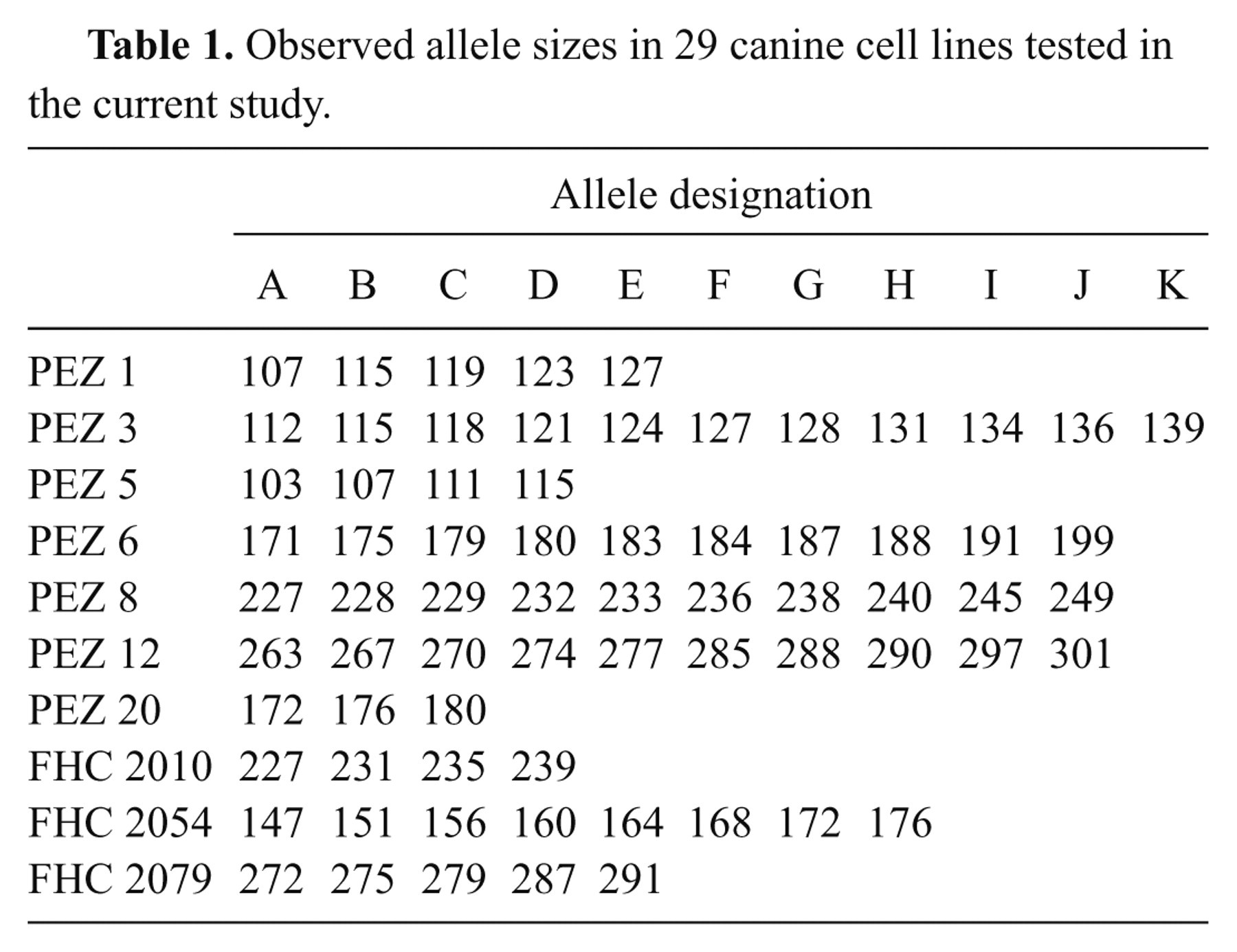
Short tandem repeat analysis of verified canine cell lines yielded 3 additional cases of mistaken identity. The STR kit tested 10 different loci; up to 11 different alleles were observed among the samples (Table 1). Alleles were designated by sequential lettering because a set nomenclature utilizing allelic ladders for canine STRs has not yet been established. This tactic allows comparisons between different cell lines or multiple samples from the same cell line to validate identity within a species. Cancer cell lines can contain many genetic alterations including loss of heterozygosity or the gain or loss of allele copies, thus, sub-lines of the same cell line may not have identical STR profiles. Considering this, looser criteria are required for the comparison of cancer cell line STR profiles. Previous work comparing STR alleles in human cell lines determined that a threshold of 75% identity was sufficient to identify all cell lines known to derive from a single source. Virally transformed and drug-resistant sublines also conformed to this threshold. 10 Cell lines isolated from different individuals showed no greater than 50% identity. 10
Observed allele sizes in 29 canine cell lines tested in the current study.
In the first case of mistaken identity, 2 samples of the widely used D17 canine osteosarcoma cell line had vastly different STR profiles (Fig. 2). In order to determine the correct STR profile, 2 additional samples of the D17 cell line were analyzed, including a sample obtained from the supplier. g Comparisons of the STR profiles between multiple samples obtained from a total of 8 osteosarcoma cell lines allowed the determination that the STR profile of one of these “D17” cell lines matched that of another canine osteosarcoma line, the Moresco line (Table 2). The other “D17” cell line matched the STR profile of the D17 sample obtained from the supplier. g In the second case, the STR profile for the canine Fitz-HSA hemangiosarcoma cell line exactly matched both early and late passage number samples from the DEN-HSA hemangiosarcoma line, indicating the 3 samples are derived from the same source. A third case of mistaken identity occurred when 2 separate samples of a putative canine melanoma cell line, CML6M, did not have similar STR profiles. However, 1 sample shared an identical STR profile with the mammary tumor cell line, CMT12, indicating contamination or mislabeling. Interestingly, the CMT12 and CMT27 cell lines had STR profiles that were 90% conserved, showing differences in only 2 alleles, indicating that they were likely derived from the same donor. This change in alleles may represent genetic drift of a single cell line or the difference between a primary tumor and a metastatic site. Additional information regarding the derivation of these cell lines or comparisons to earlier passages will help to resolve these questions. The other CML6M sample had a unique STR profile when compared to all the canine cell lines tested to date, and is presumed to be correct.
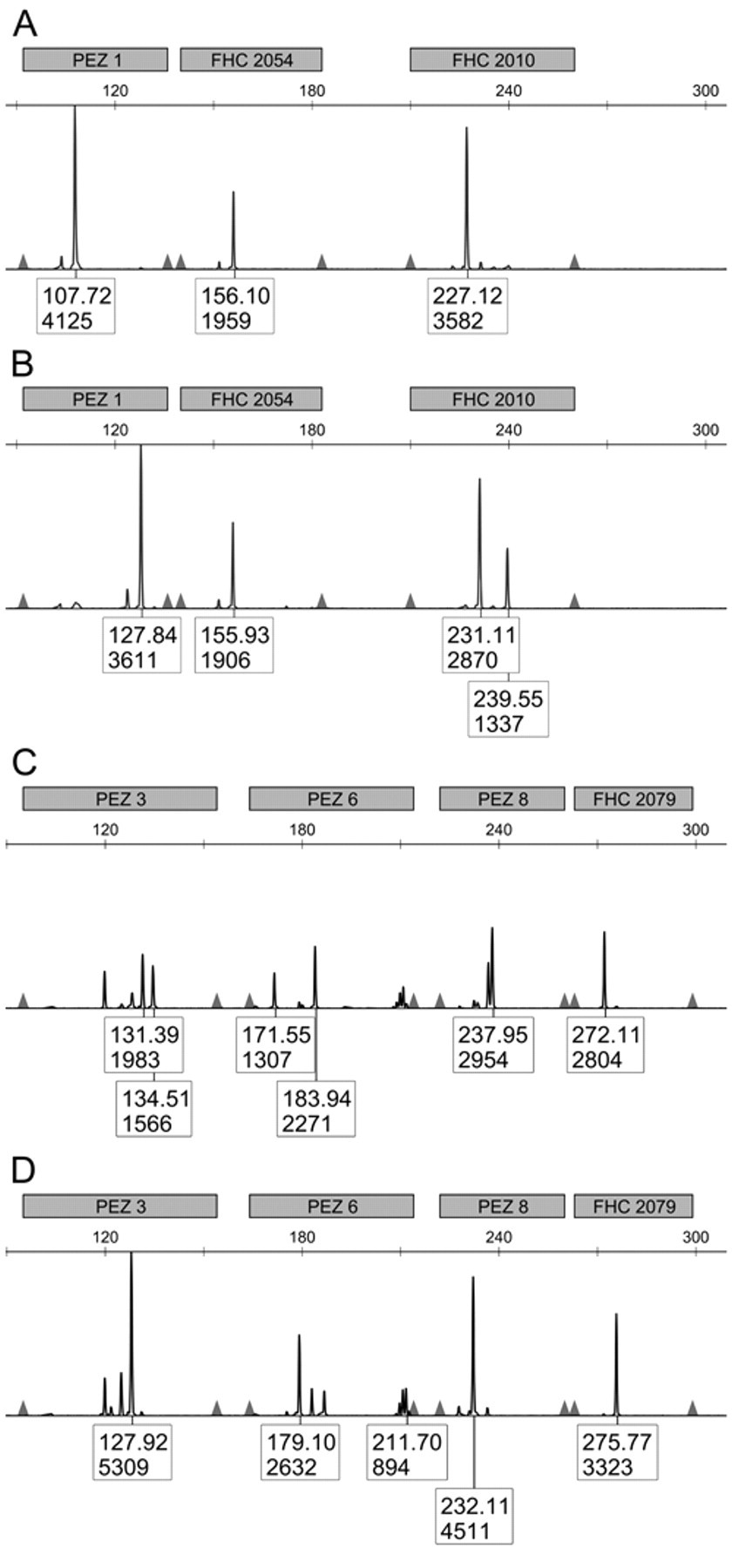
Abbreviated short tandem repeat (STR) profile of 2 cell lines originally presumed to both be D17. (
Allele distribution in 29 cell lines as determined by STR analysis.*
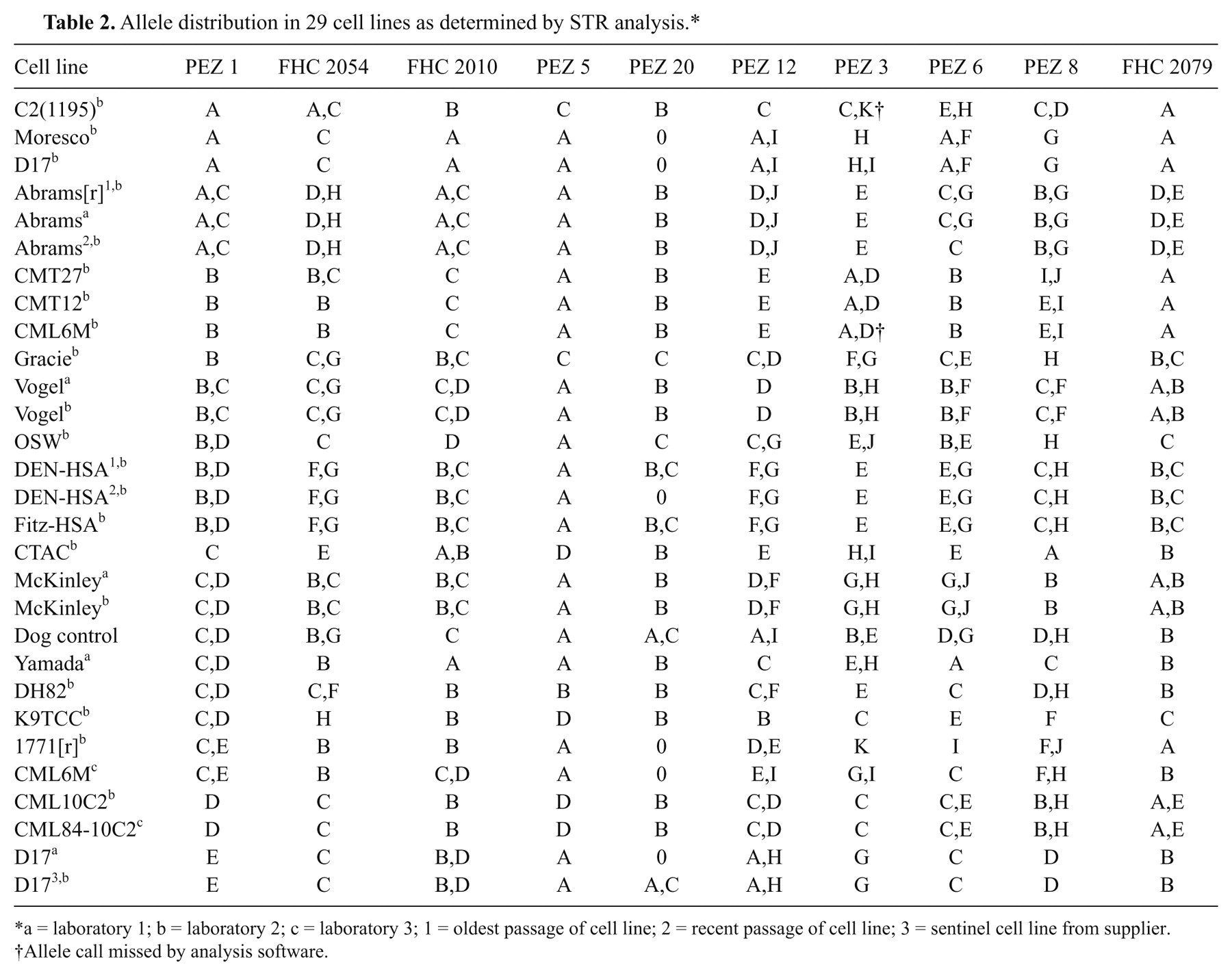
a = laboratory 1; b = laboratory 2; c = laboratory 3; 1 = oldest passage of cell line; 2 = recent passage of cell line; 3 = sentinel cell line from supplier.
Allele call missed by analysis software.
Short tandem repeat profiling is also useful for the detection of genetic drift among different passages of the same cell line. In the current study, one such case of genetic drift was observed. Comparison of 3 Abrams canine osteosarcoma cell line samples indicated that 1 later passage sample had lost one PEZ 6 allele resulting in 95% identity with the other 2 samples (Fig. 3). In all, 7 of 60 cell lines tested (12%) were misidentified or altered. Although this is lower than suggested averages, 9 it still validates the necessity of good cell culture practices including periodic cell line validation. The observed misidentifications may be due to contamination or accidental mislabeling. Thus, it is imperative to practice excellent cell culture techniques and regular testing to prevent cases of misidentification from wasting valuable time and money.
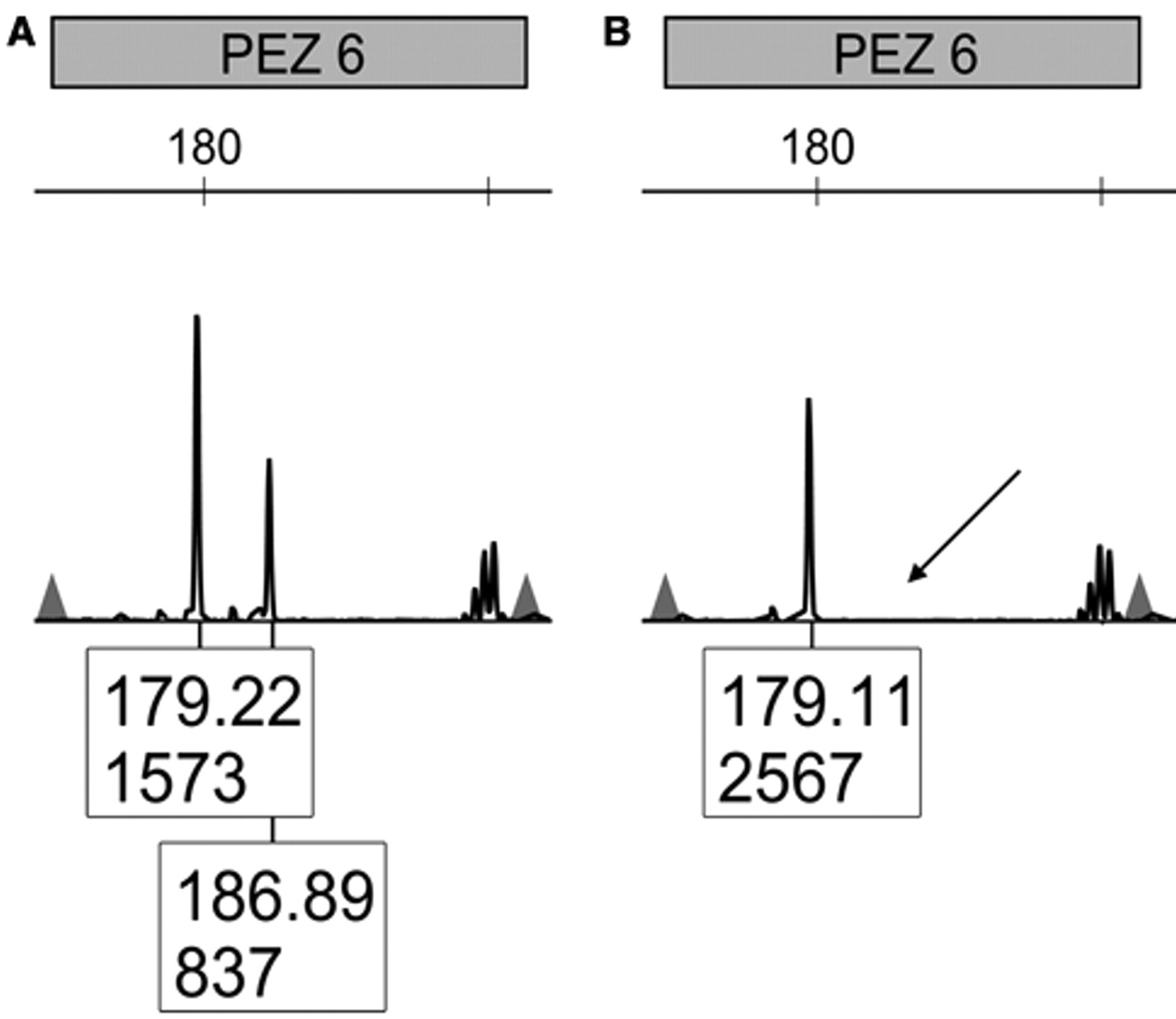
PEZ 6 locus of early (
Multiplex PCR testing using species-specific primers is a simple and inexpensive way to begin the validation process in any facility where multiple species of cell lines are used. Following species validation, PCR-based STR profiling is the current “gold standard” for cell line validation within a species. Commercial microsatellite kits are not currently available for all species. However, the increased use of STR analysis in forensics and parentage testing for breed registries has resulted in the development of STR panels for a growing variety of species. 2,3,8,12–16 Both forensic and parentage testing require the STR panels to be sufficiently complex to allow for the identification of individuals in a population and within specific breeds. The selected loci must also exhibit efficient, repeatable amplification in and between laboratories. Typically, tetranucleotide repeats have proven to be the least susceptible to error. 13 The STR panel used in the present study exceeded 99% power of exclusion for canine parentage verification in 61% of the breeds tested in a previous study. 5
For the effective development of searchable databases, a standardized nomenclature for a consistent set of microsatellite markers is required. For such nomenclature to be developed, allelic ladders for each locus must be generated so that allele size can be standardized across different facilities. As these are not currently available for the canine species, the allele sizes presented in Table 1 are specific to the capillary electrophoresis instrument and methodology used at CSU. The control canine DNA (Table 2) that was included with the STR kit may, to a limited extent, be used as an inter-facility reference point. However, the control DNA is not sufficiently precise to supplant an allelic ladder because DNA migration in capillary electrophoresis is dependent upon both size and sequence. Thus, the use of an allelic ladder would greatly enhance size measurement accuracy. 13
The average standard deviation in allele size for the control used in the current study was ±0.176 bp between runs indicating that there was a high degree of repeatability when the same instrument was used. However, different instruments and polymers may result in as much as a 4-bp shift. As a consequence, most laboratories conducting parentage testing require that the samples for offspring and all possible parents are tested concurrently to minimize error. The consistency in allele size between runs suggests that this will not be a problem for cell line validation within CSU’s core facility; however, control samples will be monitored to detect deviations in the observed allelic sizes.
Standard cell culture practices that should be implemented to avoid contamination include maintenance of separate culture media stocks for each cell line, never having multiple cell lines in the hood at the same time, and thoroughly cleaning the hood and associated cell culture equipment between cell lines. Beyond these basic practices, validation of cell lines before beginning a new study is highly recommended. To further the precision of cell line validation, it is recommended that, whenever a new cell line is cultured from tissue, a sample from the tissue donor be analyzed with STR profiling so that all future passages of the resulting cell line may be compared against the donor’s profile. Furthermore, to avoid genetic drift, it is advantageous to maintain low passage number stocks to which an experimenter can return when cultured cell lines reach a high passage number or show evidence of genetic drift on STR analysis.
Despite these current limitations, STR analysis provides a simple, inexpensive method to validate cell line consistency and identity. The development of a database of STR profiles and comparison of cell line profiles from multiple sources including early passage samples or donor tissues will improve the quality, consistency, and validity of research studies utilizing canine cell lines.
Footnotes
Acknowledgements
The authors would like to thank Jessica Prenni at CSU’s Proteomics and Metabolomics Core facility for assistance with the canine STR analysis.
a.
DNeasy, Qiagen Inc., Valencia, CA.
b.
GoTaq DNA Polymerase, Promega Corp., Madison, WI.
c.
Integrated DNA Technologies Inc., Coralville, IA.
d.
StockMarks for Dogs: Canine Genotyping Kit, Applied Biosystems, Foster City, CA.
e.
Genetic Analyzer 3130xl, Applied Biosystems, Foster City, CA.
f.
GeneScan 500 ROX Size Standard, Applied Biosystems, Foster City, CA.
g.
American Type Culture Collection, Manassas, VA.
The Molecular Oncology Laboratory at the CSU Animal Cancer Center in collaboration with the Proteomics and Metabolomics Core will facilitate species verification and canine STR analysis as a fee-for-service for interested investigators. Contact the authors for additional information.
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was funded by a grant from the Colorado State University Cancer Supercluster Program.