
Abstract
Multilocus variable number tandem repeat analysis (MLVA) has recently emerged as a genotyping method that is both robust and highly discriminatory for the differentiation of Mycobacterium tuberculosis complex (MTBC) strains, including Mycobacterium bovis. However, MLVA assessment of M. bovis isolates recovered from animals in North America has been limited. Using an epidemiologically diverse set of 41 North American M. bovis animal isolates, MLVA, based on 27 published variable number tandem repeat (VNTR) loci, was evaluated. Nineteen loci displayed polymorphism, which resulted in differentiation of 21 unique MLVA genotypes. A subset of 6 loci differentiated the isolates into 14 genetically related groups that displayed remarkable concordance with the epidemiological data gathered via traditional trace-back methods. In most cases, MLVA exhibited greater resolution than spoligotyping, which differentiated the isolates into 11 groups. MLVA genotyping of M. bovis shows great potential as a molecular typing tool for characterizing the epidemiology of M. bovis animal infections in North America. However, the greatest resolution was achieved by using a combination of both MLVA and spoligotyping.
Keywords
Introduction
In 1917, the U.S. Department of Agriculture initiated the Cooperative State-Federal Tuberculosis Eradication Program, with the goal of eliminating the risk of zoonotic transmission of bovine tuberculosis. The incidence of infection has been greatly reduced, with identified outbreaks ranging from 4 to 9 affected herds per year in domestic livestock (Dutcher M: 2007, Status of the State and Federal Cooperative Bovine Tuberculosis (TB) Eradication Program fiscal year 2006. In: Proceedings of the one hundred and tenth annual meeting of the United States Animal Health Association, pp. 683–689. USAHA, Minneapolis, MN). Potential reservoirs for these outbreaks include infected imported livestock, identified wildlife reservoirs of white-tailed deer (Odocoileus virginianus) in Michigan, and other potential residual reservoirs in livestock and wildlife. 14 To successfully accomplish the goal of eradication, robust molecular typing methods are needed to assist the epidemiological detection of reservoirs of infection, establishment of routes of transmission, and definition of at-risk populations of animals.
Molecular typing of bacterial isolates based on polymorphisms in genomic DNA (genotyping) provides a powerful approach for distinguishing Mycobacterium bovis strains and may provide valuable insight into the importance of different hosts and geographic regions in the maintenance and transmission of infection. 7 , 8 Restriction fragment length polymorphism (RFLP) typing is performed by the digestion of isolated DNA followed by probing with the insertion sequence (IS) 6110, which has been considered the definitive method for genotyping of Mycobacterium tuberculosis complex (MTBC) strains 13 ; however, for strains carrying low copy numbers of the IS6110 element, such as M. bovis, it offers limited discrimination. 7 , 8 Spacer oligonucleotide typing (spoligotyping) detects polymorphisms among strains based on the number and identity of nonrepetitive spacer elements located in the direct repeat (DR) region in MTBC strains. 4 Spoligotyping is employed as a secondary typing method to IS6110 RFLP to decrease the number of falsely clustered isolates and is widely used for genotyping M. bovis because of the low copy numbers of the IS6110 element; however, this method relies on variation found at a single genetic locus. 15 MLVA identifies polymorphisms based on the copy number of VNTRs found at multiple loci along the bacterial chromosome. MLVA is increasingly used to solve epidemiological questions associated with MTBC and is more discriminatory than spoligotyping for M. bovis in isolates found in Ireland and Chad. 9 , 15 In a recent study of M. bovis isolates found in Belgium, MLVA was highly consistent among isolates with known epidemiological links, and it discriminated better than both IS6110 RFLP and spoligotyping among isolates with no known epidemiologic links. 1
Variable number tandem repeat (VNTR) loci examined in 41 North American isolates of Mycobacterium bovis, alleles identified, and diversity indices.
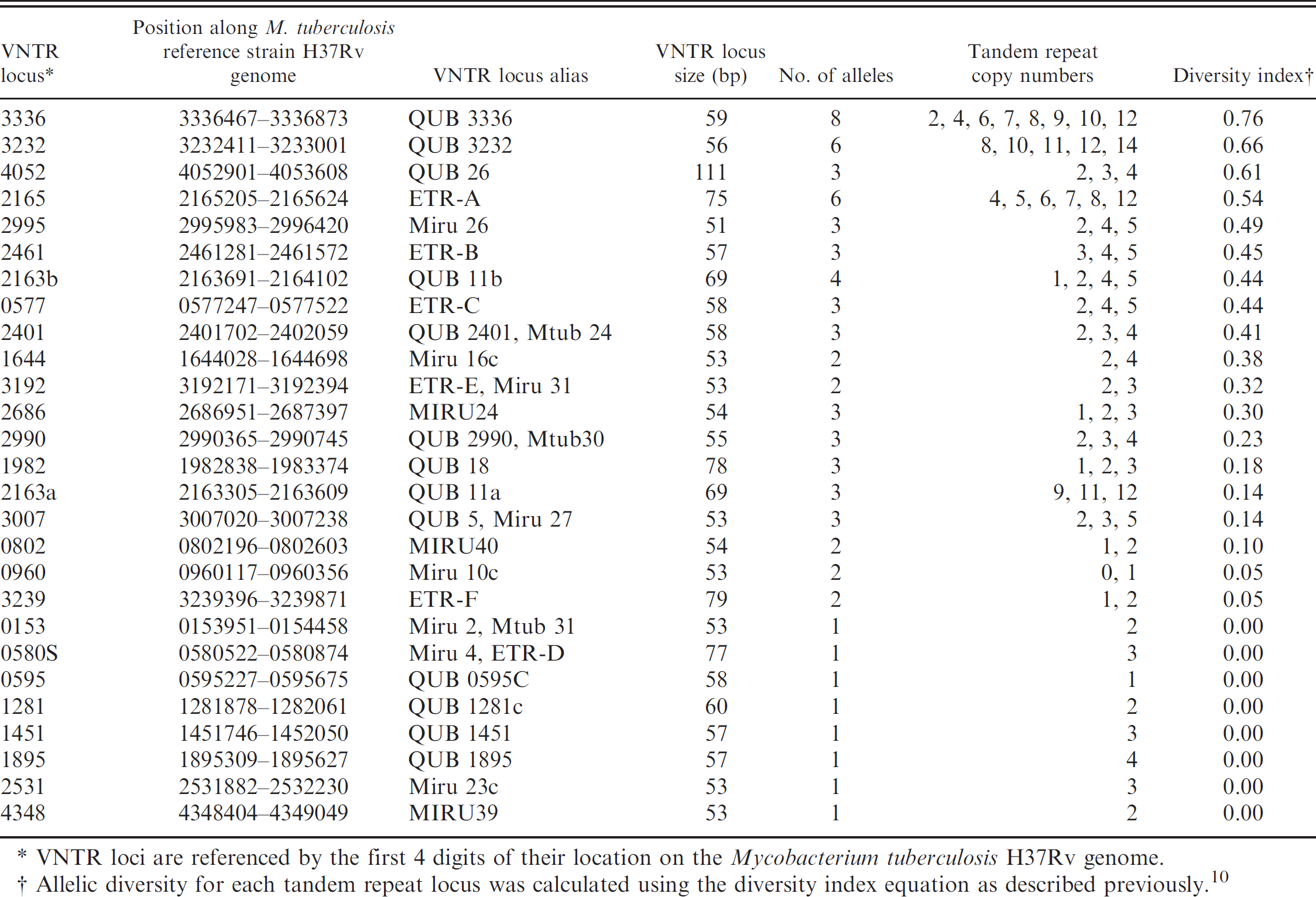
VNTR loci are referenced by the first 4 digits of their location on the Mycobacterium tuber culosis H37Rv genome.
Allelic diversity for each tandem repeat locus was calculated using the diversity index equation as described previously. 10
Assessment of MLVA of M. bovis isolates found in North America is limited. The MLVA loci that prove to be the most useful for genotyping isolates may vary from region to region and should be chosen experimentally for the target region. 15 Because this method has been shown to be a robust epidemiolog-ical tool for distinguishing M. bovis in other regions, MLVA and spoligotyping were evaluated in this study using an epidemiologically diverse set of North American M. bovis animal isolates.
Materials and methods
Tandem repeat loci
Microbiology and microbial genetics literature was reviewed to identify primer sequences for MTBC VNTR loci with potential for discriminating M. bovis strains. Thirty-five polymorphic tandem repeat (TR) loci, ranging in size between 50 and 111 base pairs (bp), were reported to have potential value. 5 , 15 , 16 , 19 After preliminary testing, 3 loci (0580, 3006, 3191) were dropped from consideration because the primers for these loci amplified the same TR locus as other primers (0580S, 3007, 3192, respectively), and 5 loci (4156, 2074, 1612, 2059, 3155) were dropped because of multiple banding patterns or no amplification of product. These problems were not resolved by adjusting polymerase chain reaction (PCR) conditions or cycling parameters. This led to the selection of 27 loci for this study (Table 1).
The genomic sequences of M. tuberculosis H37Rv and M. bovis AF2122/97 were searched to obtain the amplicon sequence for each locus. 3 , 6 The amplicons were further characterized using a TR finder program for visualization of TRs within each amplicon. 2 This information was useful for interpretation of results from PCR and for comparison of sequenced products to the published genome to validate that the TR copy numbers were accurately estimated.
Bacterial strains and DNA preparation
Forty-one M. bovis isolates were selected based on diversity of spoligotyping patterns and geographic location throughout North America. They represented isolates of wildlife origin from Hawaii, Michigan, and Montana; infected herds from California, Michigan, Minnesota, New Mexico, Texas, and Chihuahua, Mexico; a cluster of isolates from a dairy heifer-raising facility; and individual slaughterhouse surveillance cases identified in the United States that are either of unknown origin or had epidemiological traces to the states of Colima, Tamaulipas, and Aguascalientes in Mexico. The isolates, or DNA preparations from the isolates, were acquired from the culture collection of the National Veterinary Services Laboratories (NVSL; Ames, IA; Table 2). The isolates were processed at Colorado State University's level III biosafety laboratory (Fort Collins, CO). From frozen stock, the bacteria were cultured by standard methods on Middlebrook 7H10 agar, a containing sodium pyruvate, b and in Middlebrook 7H9 broth. c Two methods of DNA extraction from the bacteria were evaluated: crude DNA was prepared using a simple boiling method, and a purified preparation was obtained using a commercial kit for DNA extraction. d
PCR amplification
Polymerase chain reaction was performed in a final volume of 25 μl; mixtures contained 20 μl of 1.1 X High Fidelity PCR Supermix, e 1 μl of each primer (forward and reverse) at a working concentration of 10 μmol, f and 2 μl of template DNA from a 50-μl DNA preparation with an approximate concentration of 10 ng/μl. Positive and negative controls were included with each PCR and consisted of the same reaction mixtures, with M. tuberculosis H37Rv reference DNA g as the template for the positive control, and no template DNA in the negative control. Polymerase chain reaction was carried out using a commercial thermal cycler h with cycling parameters as described. 15 The PCR products were electrophoretically separated on 1.5% agarose gels i in a 1X Tris-boric acid ethylenediamine tetra-acetic acid (EDTA) buffer j ; product sizes were estimated using 100 and 100-plus bp DNA ladders. k
Detection and analysis of VNTRs
The numbers of TRs for isolates at each locus were determined on the basis of the number of whole repeats that could be present in a PCR product of the size estimated from the gel. Polymerase chain reaction assays for the 27 chosen loci were repeated and compared within and between gels to ensure consistent estimation of size and TR copy number.
Selected PCR products were sequenced using a commercial genetic analyzer l to verify that the amplified products contained target TR sequences and that TR copy numbers estimated from the agarose gel migrations were correct. Allelic diversity for each TR locus was calculated using the diversity index equation. 10 Tandem repeat copy numbers at each locus for each isolate were subjected to maximum parsimony analysis using phylogenetic software. 20 Additional empiric analysis was performed to determine the minimal subset of VNTR loci required to achieve the greatest resolution of the 41 isolates in the current study. Epidemiological data gathered from traditional trace-back methods were then revealed and were compared with the results from MLVA (Table 2).
Spoligotyping
DNA was extracted from the M. bovis field strains using previously described procedures. 21 Amplification of the direct variant regions for spoligotyping was performed essentially as described previously. 11 Briefly, template DNA was amplified with 2.5 units of DNA polymerase m in a 50-μl PCR mix containing 10X reaction buffer (100 mmol of Tris-hydrochloride [HCL], pH 8.3; 500 mmol of potassium chloride [KCl]; 15 mmol of magnesium chloride [MgCl2]; 0.01% [w/v] gelatin), 0.2 mmol of each deoxynucleotide triphosphate (dNTP), 1.25 mmol of MgCl2, and 20 pmol of each primer (biotinylated DRa 5′-GGTTTTGGGTCTGACGAC-3′ and DRb 5′-CCGAGAGGGGACGGAAAC-3′). Thermo-cycling conditions consisted of a 10 min denaturation step at 96°C, followed by 20 cycles of denaturation at 96°C for 1 min, annealing at 55°C for 1 min, and extension at 72°C for 30 sec, followed by a final extension at 72°C for 5 min. The PCR products were then denatured at 99°C for 10 min and applied to a nylon membrane containing 43 covalently bound spacer sequences n using a miniblotter apparatus. o Hybridization signals were detected using the chemilumi-nescent detection liquid p electronically captured, digitized, and normalized for analysis using a gel documentation system. q Interpretation of spoligotype patterns and assignment of octal codes were done as previously described. 4
Results
Genotyping of the 41 M. bovis isolates based on 19 polymorphic loci identified 21 unique MLVA genotypes (Table 2). These genotypes were further grouped into 14 types (A-N) allowing for polymorphism within 1 locus within each MLVA type (Table 2). Data are not shown for 8 of the 27 loci initially selected for characterization (0153, 0580S, 0595, 1281, 1451, 1895, 2531, 4348). These 8 loci were found to be monomorphic in this sample of 41 M. bovis isolates. Tandem repeat alleles found in the 41 isolates of M. bovis and the diversity indices of the 19 VNTR loci are presented in Table 1. Allelic diversity indices of the 19 polymorphic loci ranged from 0.76 for VNTR 3336, which had 8 alleles, to 0.05 for VNTR 3239, which had 2 alleles. A representative agarose gel is shown in Figure 1.
MLVA types A-E were comprised of single isolates with unique genotypes. Types F-J contained multiple isolates that were identical across all loci within each type. Isolates in types K-M comprised multiple isolates that were identical in all but 1 locus within each type. In each case, the difference was a single-step change in the TR copy number. Type N was a more heterogeneous group. Two of the 6 isolates (02–4170, 04–2067) were identical across all loci, with 4 isolates (04–8012, 02–5651, 02–6570, 02–1652) each varying from the 2 identical isolates at a single locus. The single locus difference for isolates 04–8012 and 02–5651 was observed at locus 3232. Isolate 02–1652 varied at locus 2461, and isolate 02–6570 varied at locus 2165.
Multilocus variable number tandem repeat analysis (MLVA), spoligotype, and epidemiological information for 41 North American Mycobacterium bovis isolates.*
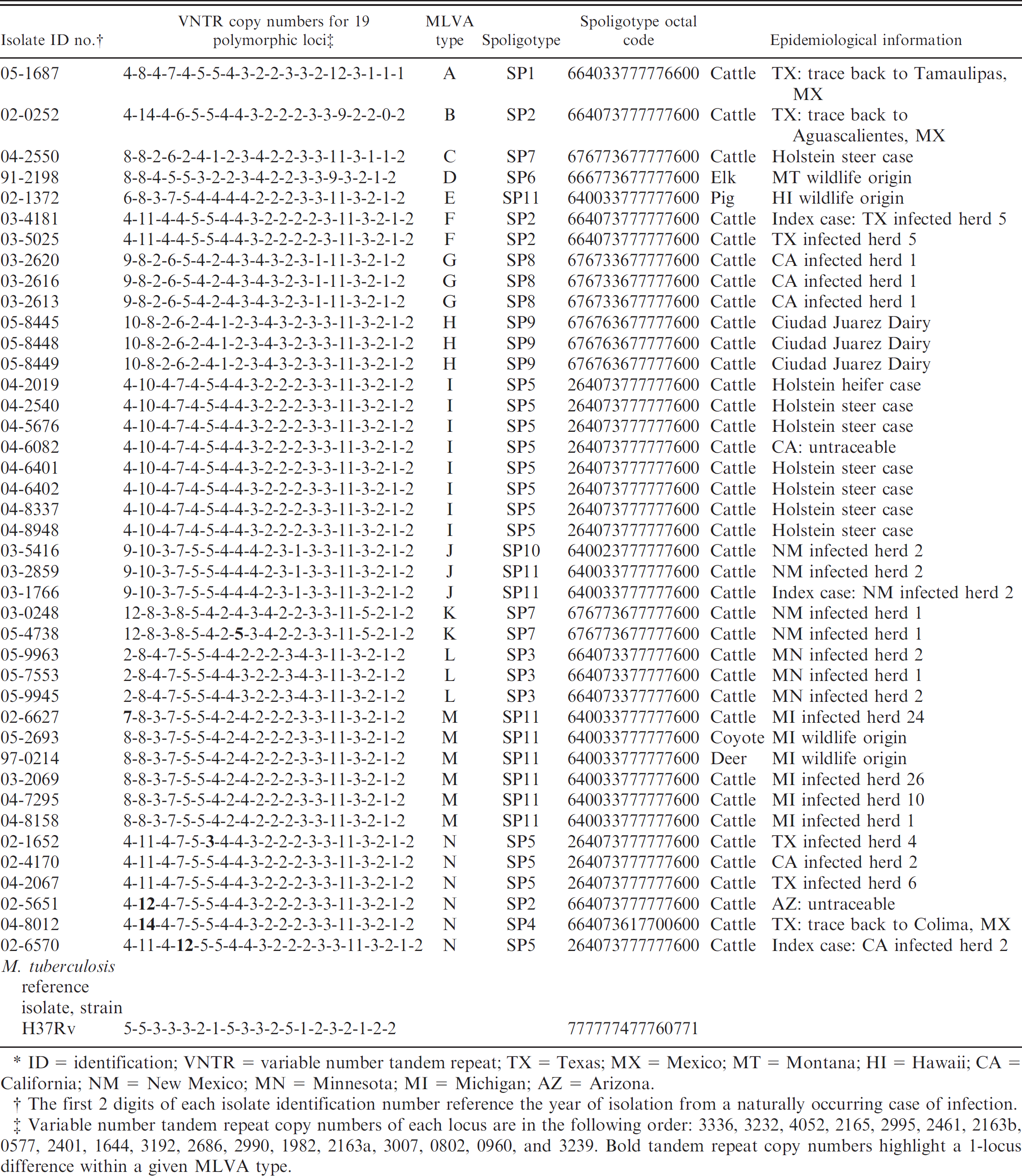
ID 5 identification; VNTR = variable number tandem repeat; TX = Texas; MX = Mexico; MT = Montana; HI = Hawaii; CA = California; NM = New Mexico; MN = Minnesota; MI = Michigan; AZ = Arizona.
The first 2 digits of each isolate identification number reference the year of isolation from a naturally occurring case of infection.
Variable number tandem repeat copy numbers of each locus are in the following order: 3336, 3232, 4052, 2165, 2995, 2461, 2163b, 0577, 2401, 1644, 3192, 2686, 2990, 1982, 2163a, 3007, 0802, 0960, and 3239. Bold tandem repeat copy numbers highlight a 1-locus difference within a given MLVA type.
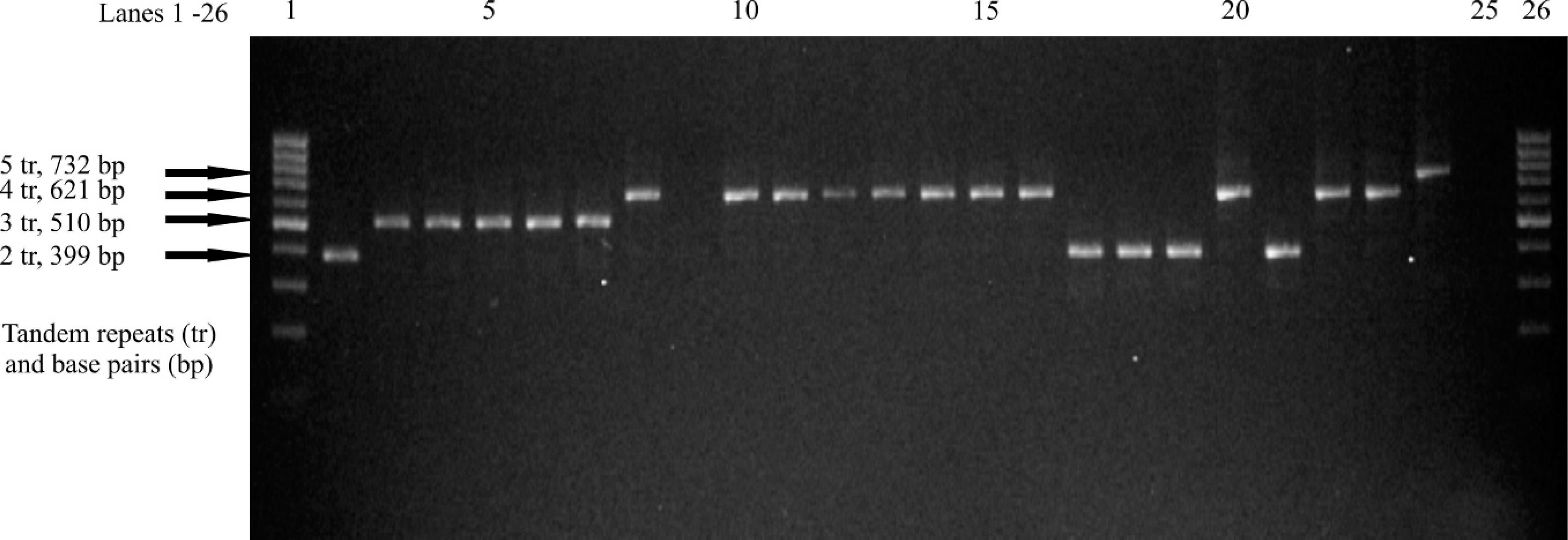
Variable number tandem repeats of 23 Mycobacterium bovis isolates at the polymorphic locus 4052, with a tandem repeat size of 111 base pairs. Lanes 1 and 26: 100 base pair ladder; lane 2: 03–2613 (California herd 1); lane 3: 04–7295 (Michigan, herd 10); lane 4: 04–8158 (Michigan herd 1); lane 5: 05–4738 (New Mexico herd 1); lane 6: 03–2859 (New Mexico herd 2); lane 7: 03–1766 (New Mexico herd 2); lane 8: 04–2019 (Holstein heifer case); lane 9: 04–2550 (Holstein steer case); lane 10: 04–2540 (Holstein steer case); lane 11: 045676 (Holstein steer case); lane 12: 04–6082 (California untraceable); lane 13: 04–6401 (Holstein steer case); lane 14: 04–6402 (Holstein steer case); lane 15: 04–8337 (Holstein steer case); lane 16: 04–8948 (Holstein steer case); lane 17: 05–8445 (Ciudad Juarez Dairy); lane 18: 05–8448 (Ciudad Juarez Dairy); lane 19: 05–8449 (Ciudad Juarez Dairy); lane 20: 02–6570 (California herd 2); lane 21: 03–2620 (California herd 1); lane 22: 02–4170 (California herd 2); lane 23: 02–0252 (Texas trace back to Aguascalientes, Mexico); lane 24: polymerase chain reaction-positive control (Mycobacterium tuberculosis H37Rv); lane 25: negative control.
Epidemiological data for each isolate are shown in Table 2. Remarkable concordance was seen between the epidemiological origin of the isolates and the MLVA types assigned. Two isolates identified from herd 5 in Texas had identical MLVA profiles and corresponded to MLVA type F. The same pattern of identity was observed within groups of isolates in a California herd (MLVA type G), in dairy herd isolates from Mexico (MLVA type H), in Holstein steer and heifer cases from a dairy-raising facility (MLVA type I), and in New Mexico herd 2 (MLVA type J). MLVA types for New Mexico herd 1 (MLVA type K), Minnesota isolates (MLVA type L), and Michigan isolates (MLVA type M) were composed of genotypes that differed at a single locus as mentioned previously.
MLVA types, MLVA fingerprints, epidemiological origin of isolates, and octal codes from spoligotyping for all 41 isolates are compared in Table 2. Spoligotyping differentiated the 41 isolates in the current study into 11 types designated SP1-SP11. In most cases, equal or better resolution of isolates was obtained with MLVA than with spoligotyping. MLVA types B and F, and 1 isolate from type N were clustered together in SP2. Similarly, 4 out of 6 isolates in MLVA type N, and all isolates in MLVA type I, were clustered together in SP5, and MLVA types K and C were clustered in SP7. All isolates in MLVA types E and M, and 2 of 3 isolates in MLVA type J, had identical octal codes (SP11), whereas, MLVA differentiates these isolates at 6 different VNTR loci. Conversely, spoli-gotyping discriminated among strains within 2 MLVA types. Spoligotyping split the 3 isolates from New Mexico herd 2 (MLVA type J), which seemed to be epidemiologically related, into 2 separate groups: 1 isolate into SP10, and 2 isolates into SP11. The 6 isolates that comprised MLVA type N were divided into 3 spoligotypes: 1 isolate into SP2, 1 isolate into SP4, and 4 isolates into SP5.
Systematic examination of various combinations of the 19 polymorphic VNTR loci was used to identify the minimal subset of loci needed to obtain maximal resolution of this group of isolates. All MLVA types could be differentiated using only 6 of the 19 polymorphic VNTR loci (4052, 2165, 2995, 2461, 3192, 2686).
Phylogenetic representation of VNTR data in the current study (41 isolates at 19 polymorphic loci) is shown as a phylogram in Figure 2. Phylogenetic clusters of closely related isolates grouped in patterns similar to MLVA types. When the minimal subset of 6 VNTR loci was used for the phylogenetic analysis, each MLVA type was identified as a distinct cluster, except type N (data not shown). This analysis must be interpreted cautiously because common epidemiological sources may not be inferred based on phylogenetic similarity without accompanying epidemiological data.
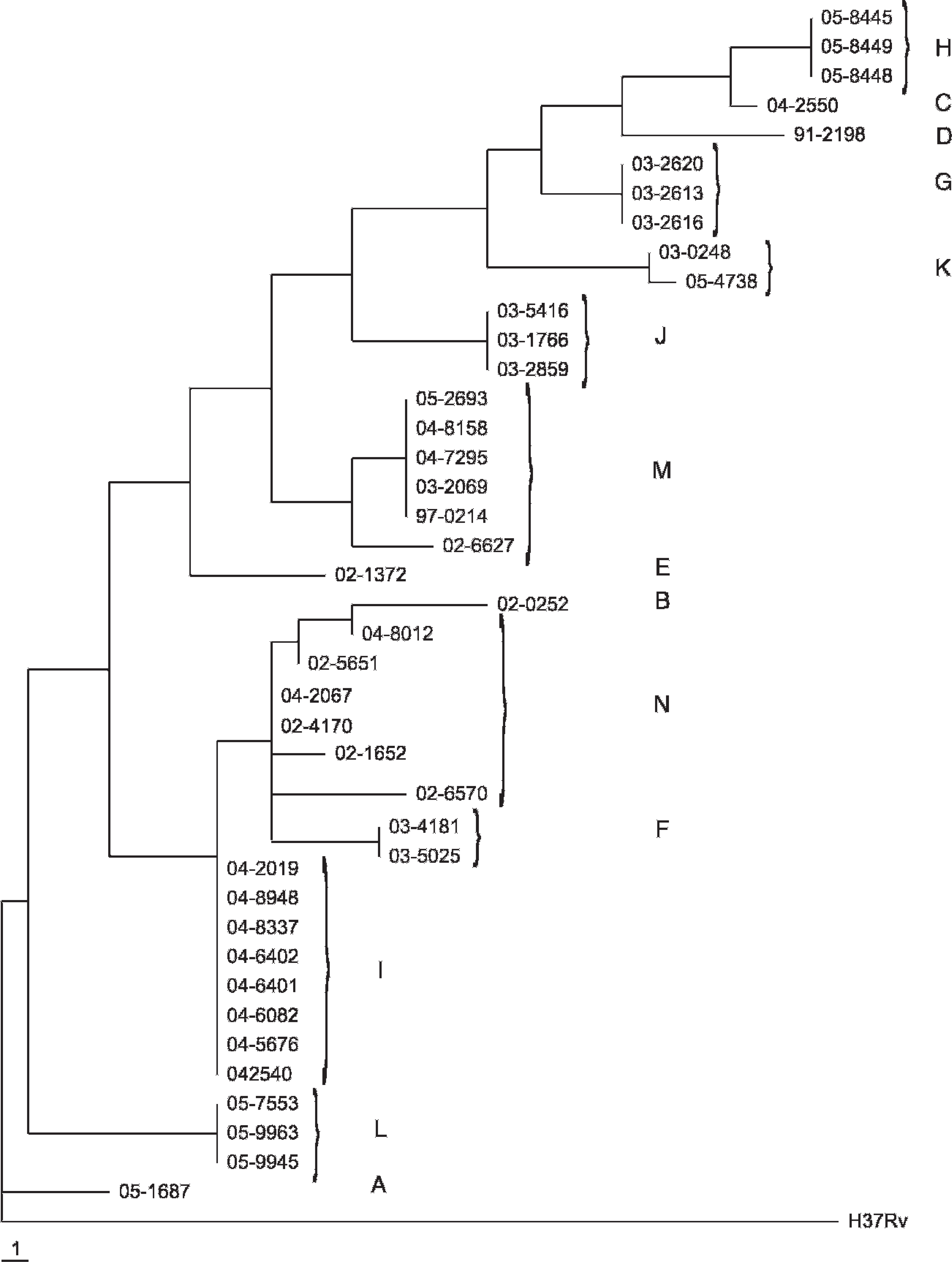
Maximum parsimony phylogram of relationships among 41 Mycobacterium bovis isolates from North America based on 19 variable number tandem repeat loci. Each step is representative of 1 tandem repeat copy number increase or decrease at a single locus. Mycobacterium tuberculosis reference strain H37Rv was used as the out-group in this analysis. Multilocus variable number tandem repeat analysis types A-N are noted.
The phylogenetic representation of the VNTR data, including the spoligotype patterns of the 41 isolates examined in this study, is shown in Figure 3. The phylogenetic tree in Figure 3 represents the genetic relatedness of the isolates but does not illustrate the distance between isolates as does the phylogram shown in Figure 2.
Discussion
In this study, MLVA was evaluated as a molecular tool for genotyping 41 North American isolates of M. bovis. The resolution of MLVA was assessed on the basis of traditional epidemiological tracing information and spoligotyping data. Six VNTR markers (4052, 2165, 2995, 2461, 3192, 2686) were identified as the minimal number of markers needed for maximal resolution of the limited set of 41 isolates screened in this study.
Ideal molecular markers must exhibit enough variability to differentiate between unrelated strains while, at the same time, demonstrate sufficient clonal stability over time to identify isolates from the same strain and trace transmission chains. 18 The significant congruence between the epidemiological origin of isolates and MLVA genotypes in the current study suggests the usefulness of MLVA as an epidemiological tool.
MLVA of the 6 isolates from Michigan highlights both the discrimination power and stability of the typing system. Michigan isolates are distinctly clustered by MLVA (type M), whereas spoligotyping included 3 other isolates in SP11 with the Michigan isolates: a wildlife isolate from Hawaii (02–1372) and 2 isolates from New Mexico herd 2 (03–2859, 03–1766). As mentioned previously, MLVA typing detected different alleles at 6 loci between the type M isolates (02–6627, 05–2693, 97–0214, 03–2069, 04–7295, 04–8158) and the three other isolates in SP11 (02–1372, 03–2859, 03–1766). Epidemiological linkage between the type M isolates and the 3 other isolates included in SP11 was not found, suggesting that MLVA typing provided greater power to resolve epidemiologically unrelated isolates than spoligotyping. The date of isolation of the Michigan isolates ranged from 1997 to 2005 (the first 2 digits of the isolate identification number denote year of isolation). MLVA fingerprints were consistent over 8 years for all isolates within type M across all loci except VNTR 3336, underscoring the stability of the alleles. The variability that occurred within type M at VNTR 3336 (D = 0.76) indicates that this marker may be too variable for applied epidemiology or that subtypes may emerge. More isolates with thorough epidemio-logical characterization need to be collected for typing to resolve the variability question. VNTR 3232 appears to provide the same challenge of hypervariability. Polymerase chain reaction amplification of these loci also required multiple tests to obtain readable results, and sequence verification was unsuccessful because of possible internal base pairing. Further analysis is necessary to determine whether these 2 loci will be sufficiently stable and reproducible to be useful epidemiological tools. Based on this sample of isolates, there would be no loss of resolution by their removal because they were not included in the subset of the 6 most informative loci that differentiated the isolates. Other research groups have experienced similar difficulties and have eliminated 1 or both of these markers from consideration. 1 , 12 , 18

Maximum parsimony phylogram with spoligotype patterns of 41 Mycobacterium bovis isolates from North America based on 19 variable number tandem repeat loci. Mycobacterium tuberculosis reference strain H37Rv was used as the out-group in this analysis.
Allowing 1 locus to remain polymorphic within each of the MLVA types did not affect the phylogenetic analysis when using the subset of 6 loci for types K, L, and M because the polymorphism within these types was observed at loci not included in the subset. In addition, each of these polymorphisms involved only a 1-step change in the TR copy number. In type N, however, the polymorphisms occurred at locus 2165 with a TR copy number change from 7 to 12, and in locus 2461, with a TR copy number change from 3 to 5. Another polymorphism was observed in the hypervariable locus 3232, which was not included in the subset. The multistep nature of these polymorphisms suggests that the isolates may be more distantly related than the isolates within other types. Although they were obtained within the southwestern United States, the origin of infection introduced into the herds was not identified, so a common ancestor could not be determined. Insight into the epidemio-logical relatedness of the isolates in type N may become clearer after additional isolates from the region are typed and/or after typing with additional polymorphic loci.
The minimal subset of 6 loci needed for the greatest resolution of the isolates in this study has been included in the optimal set of markers for differentiation of M. tuberculosis and M. bovis recommended by several other research groups. 1 , 9 , 12 , 12 . A previous study, 18 based on a worldwide collection of M. tuberculosis isolates, proposed an optimized set of 24 VNTR loci, which included a subset of 15 discriminatory loci, for use as a first-line typing method. However, the determination of which markers and groups of markers will prove to be the most useful for local application should be determined empirically. 16 Tandem repeat loci that exhibit high allelic diversity are not always the most informative. As evidence of this, VNTR 2686 has a low discrimination index (D = 0.30) and is only included in an optimal group of markers by one other research group 16 but was instrumental in the discrimination of the Minnesota isolates (05–7553, 05–9945, 05–9963; MLVA type L) from all others. With an increased number of isolates and analysis of more VNTR loci, stronger evidence for phylogenetic and epidemiologic relationships may become apparent.
Although a minimal subset of 6 VNTR markers conferred maximal resolution in this study, the sample size was limited. Both the number of isolates and the number of markers should be substantially increased to detect all possible variations and to choose an optimal set of VNTR markers for the evaluation of M. bovis found in North America. A larger database of MLVA typed isolates, in combination with spoligotyping and conventional epidemiological investigations, can provide a better understanding of transmission and distribution of M. bovis infection in North America, which will permit more precise targeting of control and eradication measures to benefit livestock industries, wildlife conservation, and public health.
Footnotes
a.
Difco™ Middlebrook 7H10 Agar, BD Diagnostic Systems, Sparks, MD.
b.
SigmaUltra Sodium Pyruvate, Sigma-Aldrich, St. Louis, MO.
c.
Difco™ Middlebrook 7H9 Broth, BD Diagnostic Systems, Sparks, MD.
d.
DNeasy® Tissue Kit, Qiagen Inc., Valencia, CA.
e.
PCR SuperMix High Fidelity, Invitrogen Corp., Carlsbad, CA.
f.
Oligonucleotides, Colorado State University, Proteomics and Metabolomics Facility, Fort Collins, CO.
g.
Mycobacterium tuberculosis genomic DNA, Strain H37Rv, Mycobacteria Research Laboratories Culture Collection, Colorado State University, Fort Collins, CO.
h.
DNA Engine Dyad® Thermal Cycler, Bio-Rad Laboratories, Hercules, CA.
i.
UltraPure™ Agarose, Invitrogen Corp., Carlsbad, CA.
j.
Promega TBE Buffer, 10X (Tris-borate-EDTA), Promega Corp., Madison, WI.
k.
GeneRuler™ 100 bp and 100 bp plus DNA Ladders, Fermentas Inc., Glen Burnie, MD.
l.
ABI Prism® 3100 Genetic Analyzer, Applied Biosystems, Foster City, CA.
m.
AmpliTaq® DNA Polymerase, PerkinElmer Life and Analytical Sciences Inc., Waltham, MA.
n.
Spacer sequences, Isogen Life Science, IJsselstein, The Netherlands.
o.
Miniblotter® 45 System, Immunetics Inc., Boston, MA.
p.
CDP-Star™, Roche Diagnostics Corp., Indianapolis, IN.
q.
ChemiDoc™ EQ System, Bio-Rad Laboratories, Hercules, CA.