
Abstract
Degradation of RNA in diagnostic specimens can cause false-negative test results and potential misdiagnosis when tests rely on the detection of specific RNA sequence. Current molecular methods of checking RNA integrity tend to be host species or group specific, necessitating libraries of primers and reaction conditions. The objective here was to develop a universal (multi-species) quality assurance tool for determining the integrity of RNA in animal tissues submitted to a laboratory for analyses. Ribosomal RNA (16S rRNA) transcribed from the mitochondrial 16S rDNA was used as template material for reverse transcription to cDNA and was amplified using polymerase chain reaction (PCR). As mitochondrial DNA has a high level of conservation, the primers used were shown to reverse transcribe and amplify RNA from every animal species tested. Deliberate degradation of rRNA template through temperature abuse of samples resulted in no reverse transcription and amplification. Samples spiked with viruses showed that single-stranded viral RNA and rRNA in the same sample degraded at similar rates, hence reverse transcription and PCR amplification of 16S rRNA could be used as a test of sample integrity and suitability for analysis that required the sample's RNA, including viral RNA. This test will be an invaluable quality assurance tool for determination of RNA integrity from tissue samples, thus avoiding erroneous test results that might occur if degraded target RNA is used unknowingly as template material for reverse transcription and subsequent PCR amplification.
Introduction
RNA is universally recognized as a more unstable molecule than DNA. RNA molecules are generally single-stranded polymers and have complex tertiary structures, making them susceptible to chemical and structural damage. 5 Moreover, RNA may be degraded by endogenous and environmental ribonucleases (RNases). 1 Degraded RNA can cause false-negative results leading to delayed diagnosis or misdiagnosis from laboratory tests, where analyses depend upon detecting RNA templates such as RNA viruses in animal tissues. Therefore, it is important to protect tissue samples intended for RNA analysis against RNA degradation. If live animals are biopsied or euthanized at the laboratory, sample degradation can be minimized with asepsis, nuclease-free instrumentation, and by using RNase antagonists and sample preservative media. However, when samples are taken remotely, particularly in the field, it is difficult to eliminate and prevent endogenous and environmental RNases. This problem is exacerbated when transport to the laboratory involves long time periods. Sample preservation media can be used; however, commercial preparations are expensive and guarantee only to delay RNA degradation for a few hours if cold temperatures cannot be maintained. Other in-house preparations are cheaper but frequently contain toxic reagents, such as ethanol and beta-mercaptoethanol, for which the International Air Transport Association stipulate specific, and often expensive, packaging and transport arrangements. Freezing the samples immediately after excision might preserve RNA in some samples, but care must be taken that the samples remain frozen at all times during transport.
In the laboratory, it is often difficult to ascertain if RNA has been degraded in a sample, whether as a result of transport, storage, or the extraction process. Denaturing gel electrophoresis with Glyoxal or formaldehyde can be performed on an RNA extract if RNase-free electrophoresis equipment, reagents, and buffers are used. Electrophoresis will show if degradation has occurred, but it typically requires large amounts (approximately 10 μg) of RNA 10 and hence is unsuitable for diagnostic purposes in which limited RNA may be obtained from specimens. In addition, RNA gels often indicate incomplete degradation, observed as a stained smear, and do not indicate if the degradation is sufficient to render the RNA unsuitable for reverse transcription and subsequent PCR amplification. Alternatively, many molecular amplification methods include a host-specific “housekeeping gene,” test but these usually determine the integrity of the sample's DNA, such as a decapod 18S gene polymerase chain reaction (PCR), 4 which will not be as susceptible to degradation as the RNA. To determine the integrity of RNA, test-specific and species-specific mRNA internal controls intended for standardization in quantitative PCR might be suitable for adaptation to internal controls for detection of intact RNA, such as porcine cytokine mRNA primers 9 might be suited as an integrity control for detection of porcine viruses. In addition, other chromosomally derived RNA species such as 18S rRNA might be used as RNA control tests. The 18S rDNA sequence has been used by many researchers in phylogenetic studies but has been found to exhibit sufficient sequence variation that different primers are normally required for different species, groups, or closely related taxa. It has even been reported that 18S rRNA may be polymorphic within the same individual animal. 2 Hence, the use of chromosomally derived templates such as mRNA or 18S rRNA would necessitate maintenance of primers and test parameters for each host species or closely related group likely to be tested in a laboratory. It would be notably simpler and more economical if a universal test could be used on multiple host species and tissues.
The method described here detects intact 16S ribosomal RNA (rRNA) transcribed in vivo from mitochondrial DNA (mtDNA). A significant degree of sequence conservation of this gene across species has long been noted 3 as mtDNA is inherited maternally only, to preserve the functionality of the RNA product. The aim of this study was to develop a method, based upon existing mtDNA amplification primers, which would detect the 16S rRNA products in animal tissue cells, irrespective of species, thus providing a universal quality assurance tool for tests based upon RNA analyses. This method will give the laboratory analyst the confidence that RNA degradation of a sample had not occurred to the extent that reverse transcription PCR-based diagnostic tests would be affected. This knowledge is important especially where transport and/or storage conditions may not have been optimal for the subsequent RNA analysis. The failure of rRNA amplification may demonstrate the unsuitability of a specimen and prevent the false interpretation of test data.
Materials and methods
Animal tissue samples. Animal tissue samples were obtained through the diagnostic activities of the Tropical and Aquatic Animal Health Laboratory (TAAHL). When fresh tissues were not available, frozen material was used where these tissues had been previously excised from freshly euthanized animals, immediately frozen, and not thawed until used for these experiments. For comparison, some tissues were placed in commercial preservative. a The range of animal tissues used is listed in Table 1.
Animal tissues used to optimize and validate reverse transcription PCR for 16S rRNA.

The commercial preservative RNAlater was used (Qiagen, Doncaster, Victoria, Australia).
Extraction of RNA. RNA was extracted from animal tissues with a commercial kit that includes enzymatic removal of DNA. b Eluates were examined for purity and concentration using ultra-violet spectrometry. Eluates from 4 different tissues (barramundi muscle, chicken spleen, bovine brain, and bovine blood) were tested using the rDNA PCR step only, described below, to confirm the absence of residual rDNA. Eluates containing RNA were stored at −80°C.
Determination of pan-specific primers. A number of reported 16S rDNA primers were compared for ability to transcribe 16S rRNA and amplify the synthesized cDNA. The primers tested were sourced from previous publications. 6,8,11 Primers were compared in different combinations, where melting temperature and directional compatibilities were noted. Transcription and amplification reaction parameters initially were determined from the enzyme requirements, primer melting temperatures, and expected amplicon sizes and subsequently were optimized experimentally to give the parameters described below. These optimum conditions were used to test all samples listed in Table 1.
Reverse transcription PCR to detect 16S rRNA. Total RNA (500 ng to 1 μg) extracted from tissues was reverse transcribed into cDNA using Moloney murine leukemia virus (M-MuLV)-derived reverse transcriptase c primed with 16S-1472table1 6 (5‘-agatagaaaccgacctgg-3’) according to the manufacturer's instructions. The resulting cDNA was stored at −20°C.
PCR mixtures were prepared from 1× polymerase buffer with ammonium salts, d 1.5 mmol 1−1 magnesium chloride, 200 μmol 1−1 each deoxynucleotide triphosphate (dNTP), 5 pmol primer 16S-1472, 6 5 pmol primer 16SL-3f 11 (5‘-aattactttagggataacagcg-3’), 5 μg bovine serum albumin (BSA), 0.5 U Tag polymerase, d 2.5 μl cDNA and sterile nuclease-free water to 25 μl. Reactions were cycled at 94°C/5 min; 40 × (94°C/30 sec; 42.5°C/30 sec; 72°C/45 sec); and 72°C/3 min. Amplicons of approximately 200 base pair (bp) were resolved by 1.5% agarose gel electrophoresis and ethidium bromide staining.
Comparison of degradation periods of rRNA and viral RNA in blood samples. Fresh bovine blood from a single healthy animal (approximately 250 ml) was collected with lithium heparin anticoagulant. Aliquots of 900 μl were aseptically transferred to sterile 2-ml tubes.
Akabane virus was cultured in baby hamster kidney (BHK) cell monolayers in minimal essential medium with 10% fetal bovine serum. Aliquots of 100 μl cell culture supernatant containing Akabane virus were aseptically added to the blood samples and mixed by inverting the tubes. Twenty spiked blood samples were stored at each of the following temperatures: 37°C, 22−25°C (ambient temperature), 4°C, −20°C, and −80°C.
Akabane virus RNA was detected in the samples with reverse-transcription (RT) PCR using primers modified from those previously described. 7 Primer sequences were AKAI206F-MOD 5‘-cacaaccaagtgtYG ATCTTA-3’ and AKAI560R-MOD 5‘-AAGTTGACATCCATYC-CATC-3’. Highlighted bases had been modified to ensure primer compatibility with multiple Akabane data retrieved from Genbank (data not shown). Reverse transcription was conducted using AKAI206F-MOD and M-MuLV c according to the manufacturer's instructions. Amplification of target cDNA was conducted in 25 μl reactions containing 1× polymerase buffer, d 1.5 mmol 1−1 magnesium chloride, 200 μmol 1−1 each dNTP, 10 pmol each primer, 10 μg BSA, 1 U Taq polymerase, d 2.5 μl cDNA, and a volume balance of sterile nuclease-free water. Reactions were cycled at 94°C for 5 mins; 40× (94°C for 30 secs; 55°C for 30 secs; 72°C for 45 secs); 72°C for 5 mins and resolved using 1.5% agarose gel electrophoresis with ethidium bromide staining. Amplicons derived from Akabane virus RNA were approximately 350 bp.
Spiked blood samples were removed from storage temperatures at time intervals from 3 hrs to approximately 2 mos, or until negative results were obtained from 3 successive samples. Total RNA was extracted as described above and tested with the rRNA RT-PCR and the Akabane-specific RT-PCR simultaneously. Unspiked bovine blood was similarly tested as a negative control for the viral test and a positive control for the rRNA test, and cell culture supernatant was tested as a positive control for the viral test. Blood samples were discarded after each test, and new aliquots were used at each testing time period.
Comparison of degradation periods of rRNA in different tissues. Brain, spleen, and liver tissue were removed immediately from a freshly euthanized barramundi (Lates calcarifer). Each tissue was aseptically subdivided into approximately 50-mg aliquots in sterile 1.5-ml RNase-free plastic tubes. RNA was extracted from 1 sample of each tissue and tested with the rRNA RT-PCR, and remaining samples of each tissue were incubated at 37°C and 25°C. One tube of each tissue was removed from each temperature after 1, 2, 4, and 7 days and tested for RNA integrity using the rRNA RT-PCR.
Results
Extraction of RNA. RNA eluates from barramundi muscle tissue, chicken spleen, bovine brain, and bovine blood showed no evidence of residual DNA when tested only with the rDNA PCR step.
Reverse transcription PCR While some primer combinations were observed to reverse transcribe and amplify only from some of the taxonomic groups tested, or amplified multiple products from some groups, cDNA primed with 16S-1472 6 followed by PCR using 16S-1472 6 and 16SL-3F 11 consistently performed equally well in the PCR with all tissues and preservation methods tested (Table 1) and was used in subsequent analyses. This primer combination reverse transcribed and amplified a region of approximately 200 bp (Fig. 1).
Comparison of degradation periods of rRNA and viral RNA in blood samples. When identical samples of bovine blood spiked with Akabane virus were tested simultaneously for rRNA and virus at timed intervals, both reactions ceased detectable amplification at approximately the same time (day 6–7) at the higher incubation temperatures (37°C and ambient). At chilled and frozen temperatures, both PCRs gave positive results through the entire duration of the experiment. These results are shown in Table 2, and a gel image of observed degradation on day 6 is depicted in Fig. 1. There were no temperature-time combinations where results differed between the 2 tests on a single sample.

Results of 16S rRNA RT-PCR performed on bovine blood stored for 6 days at various temperatures. M = 100 bp DNA laddere; lane 1 = negative control RT-PCR; lanes 2 to 5 = RT-PCR from blood stored at 37°C (very weak reaction), ambient (weak reaction), 4°C and −20°C (positive reactions), respectively; lane 6 = negative control PCR.
Comparison of degradation periods of rRNA in different tissues. These results are depicted in Table 3. After 1-day storage at ambient and higher temperatures, piscine spleen, liver, and brain samples were visibly deteriorating, yet rRNA was amplified from all 3. By 2 days, the spleen stored at 37°C had liquefied and no rRNA was detected, and a replicate sample stored at ambient temperature showed visibly weaker amplification (data not shown). At ambient temperature, spleen tissue RNA could no longer be reverse transcribed by the fourth day, and liver tissue RNA showed weaker amplification at this time, while RNA from brain tissue retained integrity for the longest duration (>7 days). Of interest, rRNA from the brain tissue retained sufficient integrity for reverse transcription for a longer period than rRNA in blood.
Comparison of degradation periods of 16S rRNA and single-stranded viral RNA (Akabane virus) in blood samples.∗
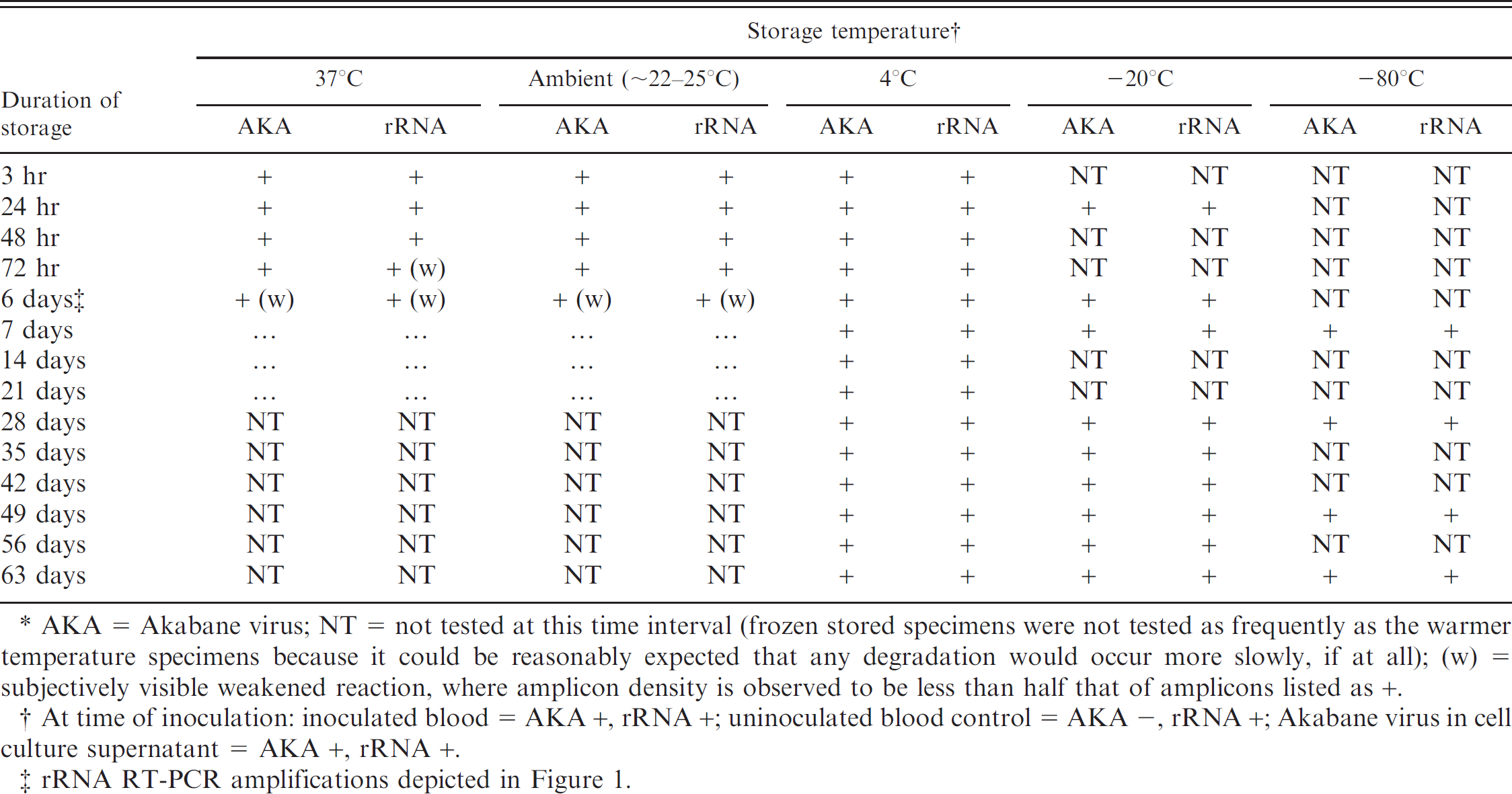
AKA = Akabane virus; NT = not tested at this time interval (frozen stored specimens were not tested as frequently as the warmer temperature specimens because it could be reasonably expected that any degradation would occur more slowly, if at all); (w) = subjectively visible weakened reaction, where amplicon density is observed to be less than half that of amplicons listed as +.
At time of inoculation: inoculated blood = AKA +, rRNA +; uninoculated blood control = AKA –, rRNA +; Akabane virus in cell culture supernatant = AKA +, rRNA +.
rRNA RT-PCR amplifications depicted in Figure 1.
Discussion
This method used established 16S rDNA primers to reverse transcribe 16S rRNA into cDNA and subsequently amplify the cDNA with PCR. The results demonstrated that rRNA can be detected in this manner from all of the animal tissue types tested if samples are fresh or sufficiently preserved to prevent RNA degradation. Testing for rRNA in replicate blood samples stored at different temperatures indicated that, at higher temperatures when tissue degradation would be expected to occur more radoily, the RT-PCR ceased detecting rRNA earlier than at the cooler temperatures. It was concluded that the repeated inability to reverse transcribe and amplify from samples stored at the higher temperatures was an indication of rRNA degradation in the blood cells. Credence is given to this conclusion upon consideration that the degrading enzymes will be more active at the higher temperatures used and that potential hemin-derived PCR inhibitors released from cell breakdown and that result in loss of amplification would be neutralized by the incorporation of BSA in the reaction mixtures.
Comparison of degradation periods of 16S rRNA in different tissue samples in laboratory conditions.∗

(w) = subjectively visible weakened reaction where amplicon density is observed to be less than half that of amplicons listed as +.
These times taken for RNA degradation should be observed for comparison of tissues only. It is not intended that these subsamples will represent postmortem samples from the field. It is likely that the lack of asepsis, the larger sample mass, movement/agitation, and the variable temperature and humidity encountered during field sampling and transport would affect the rate of RNA degradation.
To confirm that rRNA degradation also would imply degradation of viral RNA in vivo, replicate blood samples were spiked with cell culture supernatant containing Akabane virus. At timed intervals, samples from various storage temperatures were tested for both 16S rRNA and Akabane RNA. At the higher temperatures, where rRNA degradation was observed, the degradation of viral RNA occurred at a comparable rate. This demonstrated that in this model at least, the rRNA degradation represented degradation of viral RNA in vivo. Chilled temperatures are recommended over freezing for storage of blood fractions containing Arboviruses (http://www.oie.int/eng/normes/mmanual/A_summry.htm), so degradation was not expected to readily occur at these temperatures in the model used. Hence, it must not be assumed that the actual degradation times determined here are representative of RNA in other tissue-virus models, and these results must be observed only for comparative purposes between the 2 RNA degradation analyses. A blood-Akabane model was used here for convenience because it was possible to prepare the replicate homogeneous samples required for a timed trial, where time and temperature were the only variables, and because this laboratory has in place an RT-PCR, which has been shown to successfully detect part of the Akabane virus RNA. It may be expected that double-stranded RNA, for example from Orbiviruses, might be more resistant to degradation.
Other tissue specimens were shown to degrade more radoily, presumably because of higher cell densities in solid tissue and subsequent amounts of endogenous RNases and proteases, which demonstrated that degradation rates of RNA vary according to the immediate environment. It must be noted that the tissues tested in this study were aseptically removed and subdivided immediately following euthanasia in the laboratory and were stored in constant temperature–controlled incubators. In the field, RNA in a tissue may be affected by environmental RNases, endogenous RNases from surrounding tissues, lack of asepsis, sample size, variable temperatures, variable humidity, and potential agitation through transport. Hence, the results presented in this study must serve only as a comparison of tissues, and not as a guide to sample “shelf-life.”
The rRNA test described herein can be used in diagnostic laboratories to examine the integrity of RNA in tissue samples, which is particularly useful for quality assurance when the transport and/or storage conditions have deviated from optimal conditions, or where suitability of the template RNA may be otherwise questioned or doubted. The ability to use a standard protocol for multiple animal types is more convenient and economical than maintaining reagents for multiple different tests. The cDNA synthesis can be carried out simultaneously with other reverse transcriptions as conditions are standard for this reaction. The PCR step can be carried out simultaneously with other reactions if a gradient block thermal cycler is used, where more than 1 primer annealing temperature can be performed at any one time, and if the primer annealing and extension durations are similar. Depending upon the reaction conditions of the specific target PCR, the tests theoretically may be suited to multiplexing, but this would require separate validation in the specific test models and was not attempted here.
The results demonstrate also that the 16S primers prime amplification from the host's mtDNA, which emphasizes the requirement to eliminate DNA from the reverse transcription template. Failure to do so may result in contaminating host DNA carry-through to the PCR reaction and give false-positive results. A DNA-free extract can be confirmed as such by performing the 16S PCR step alone on an extract, where amplification will indicate the presence of rDNA.
However, because the 16S rDNA region can be amplified directly by the same primers, the primers may be used also to determine DNA template integrity for direct PCR tests such as detection of DNA virus or bacterial genes in animal tissues, without the reverse transcription step needed for RNA templates. In the event that an animal tissue requires detection of both RNA and DNA templates, the total nucleic acid extraction will need a subsample to be treated with DNase prior to testing for RNA integrity.
Acknowledgements
Samples for this project were collected by DPI&F veterinarians Drs. Rebecca Taylor, Ian Anderson, and Glen Edmunds. Akabane virus was cultured and provided by Dr. Nick Moody and Mr. John Hoad (TAAHL, DPI&F). Technical assistance was provided by Ms. Kelly Condon and Mr. Christopher Wright (TAAHL, DPI&F)
Footnotes
a.
RNAlater; Qiagen, Doncaster, Victoria, Australia.
b.
Wizard SV Total RNA Isolation System; Promega, Annandale, New South Wales, Australia.
c.
Revert Aid; MBI Fermentas, Quantum Scientific, Murarrie, Queensland, Australia.
d.
Taq Polymerase (recombinant); Quantum Scientific, Murarrie, Queensland, Australia.