
Abstract
A real-time reverse transcriptase (RT)-PCR assay, applying light upon extension (LUX) fluorogenic primers, was developed for rapid and efficient detection of Newcastle disease virus (NDV). The method, which targets the fusion (F) protein gene of the viral genome, gave positive signal with all NDV isolates tested (32/32), while negative results were obtained with heterologous pathogens (35/35), including 13 avian influenza virus isolates. The detection limit of the assay was approximately 10+1.2 egg infectious dose (EID)50/0.2 ml and 10+2.2 EID50/0.2 ml for virus suspensions and spiked chicken fecal samples, respectively. As expressed in plasmid copy number, the procedure has a sensitivity of approximately 20 copies of the plasmid harboring the target gene. Due to its high specificity, sensitivity, and relative simplicity, the LUX RT-PCR assay provides a novel, rapid, and practical tool for the detection of NDV.
Newcastle disease (ND) is one of the most devastating diseases of domestic birds and it poses a considerable economic threat to the poultry industry worldwide. The disease is caused by Newcastle disease virus (NDV), also known as avian paramyxovirus 1 (APMV-1), a member of the Avulavirus genus within the Paramyxoviridae family. The virus has a wide host range; birds belonging in 27 orders have been reportedly affected by the disease. 7 The severity of clinical signs varies among species, the mortality rate being the highest in chickens. 7 Depending on their virulence properties, NDV strains are divided into 3 pathotypes: lentogenic, mesogenic, and velogenic; clinical signs in infected birds range from inapparent infection to acute disease and high mortality. 1
The official method for the diagnosis of ND—recommended by the Office International des Epizooties—is the isolation of NDV using embryonated chicken eggs and subsequent characterization of the virus isolate by hemag-glutination inhibition assay (HI). 1 This conventional procedure takes at least 4 to 7 days to accomplish. The clinical, economical, and epidemiological needs to rapidly identify NDV infection have led to the development of several PCR-based assays, including real-time PCR methods using SYBR Green and TaqMan technologies. 8,12,15 However, these technologies have some limitations: 1) in a SYBR Green-based assay, nonspecific priming events may result in inefficient amplification of expected products and a wide variation in sensitivity (from 10 to 1,000 copies) in the dynamic range 3,9 ; and 2) in the TaqMan system, although the use of 3 oligonucleotides provides high specificity, the risk of having mismatch(es) between the target/probe nucleotide sequences may lead to false negative results, especially considering the genetic variations in field isolates. 14
In order to provide an alternative molecular approach for the improved detection of NDV and to overcome the above-mentioned problems, a novel real-time reverse transcriptase (RT)-PCR assay that uses light upon extension (LUX) fluorogenic primer was developed. The LUX primer set includes a fluorogenic primer with a fluorophore attached to its 3′ end and an unlabeled primer (http://www.invitrogen.com/lux). a The fluorogenic primer has a short tail sequence of 4 to 6 nucleotides on the 5′ end that is complementary to the 3′ end of the primer. The resulting hairpin secondary structure provides optimal quenching of the fluorophore. When the primer is incorporated into double-stranded DNA during PCR, the fluorophore is dequenched and the signal increases by up to 10-fold. Thus, unlike in TaqMan PCR, LUX primers do not require special probes or quenchers. Furthermore, this setup enables the melting curve analysis of PCR products, providing a convenient way of differentiating amplicons from nonspecific and primer-dimer artifacts.
List of NDV strains (vaccines/field isolates) investigated by the LUX real-time RT-PCR and their results in the specificity test.

ch = chicken; Hu = Hungary; V = velogenic; Nd. = not determined; L = lentogenic; tk = turkey.
Heterologous avian pathogens investigated by the LUX real-time RT-PCR and their results in the specificity test.

ch = chicken; dk = duck; Hu = Hungary; md = muscovy duck; gf = guinea fowl; tk = turkey.
A collection of 32 archived NDV isolates of different origin, representing 7 genotypes of the described 8, 5 3 NDV vaccine strains, and 35 heterologous pathogens including 13 avian influenza virus (AIV) strains and 6 avian paramyxovirus strains (Tables 1, 2) was tested to evaluate the specificity of the assay. The archived samples and isolates were collected from clinical samples in the Virology Department of the Central Veterinary Institute, Debrecen, Hungary, from 1969 to 1990, or were obtained from Dr. B. Lomniczi (Veterinary Medical Research Institute of the Hungarian Academy of Sciences, Budapest, Hungary), and CEVA Phylaxia Ltd. (Budapest, Hungary), b and they tested positive for the presence of the respective pathogens by using standard diagnostic procedures at the authors' laboratory. Virus samples were identified by propagation in embryonated chicken eggs and hemagglutination (HA) test according to the World Organisation for Animal Health (Office International de Epizooties) manual. 1 Isolated viruses were passaged in embryonated chicken eggs to make stock solutions, and the titer of virus suspension was determined as the 50% egg infectious dose (EID50).
LUX primers specific for a conserved region of the fusion (F) protein gene of NDV were designed using sequence data available in the GenBank (www.ncbi.nlm.nih.gov). For a comparison of nucleotide sequences derived from isolates representing various geographic origins and different periods of time, alignments were prepared by using GeneDoc software. 11 Oligonucleotide primers were designed using the Primer Designer software Version 2.0, c and they were later modified to meet the requirements for LUX RT-PCR assays. 10 The 2 oligonucleotides used in the assay, ND-703F-JOE 5′- catctt AGT GGC AGT TGG GAA GAT G-3′ (the T next to the 3′ end G is labeled with the fluorophore JOE, 6-carboxy-4,5-dichloro-2,7-dimethoxyfluorescein; lowercase letters indicate the nucleotides added to the target-specific sequences to form hairpin), and ND-845R 5′- GTG GYC CGA ATA CTG TAG-3′ (Y = T/C) were flanking a 143-nucleotide-long fragment of the F protein gene. The locations of the forward and reverse primers on the SL03 isolate (GenBank accession number DQ228922) are 528–546 and 670–653 nucleotide positions, respectively.
RNA extracts, prepared from 140 μl of sample (allantoic fluid, tissue homogenate, or spiked fecal sample) by using the QIAamp Viral RNA Mini Kit as recommended by the manufacturer, d were eluted in 70 μl of elution buffer.
One μl RNA per reaction was used as template for the ensuing One Step RT-PCR assay d in a final volume of25 μl and by applying the following conditions: 1 μl of kit-supplied enzyme mixture (including reverse transcriptase and hot-start Taq polymerase), 0.6 μM of forward and 1 μM of reverse primer, 400 μM (each) deoxynucleoside triphosphate, 1 mM MgCl2, 5 U of Rnase inhibitor, e 5 μl 5× reaction mix, and 14.2 μl Rnase-free distilled water. The RT step conditions were 60 minutes, 50°C, and 15 minutes, 95°C. The PCR cycling protocol was as follows: 45 cycles of 94°C, 15 seconds; 58°C, 35 seconds; and 72°C, 30 seconds, followed by 72°C, 1 minute. Dnase/Rnase-free distilled water served as negative control in each reaction.
After PCR, melting curve analysis of the amplified PCR products was carried out from 48°Cto96°C in 0.5°C/10 sec increments. In order to test its versatility, the assay was tested on several instruments including iCycler, f ABI 7500 real-time PCR system, g and RotorGene 3000, h by using the same reaction conditions, including the thermal profiles, as described above.
The amplified DNA fragments were purified by using the QIAquick PCR purification kit c according to the manufacturer's instructions. The nucleotide sequence of an NDV amplicon was determined by using the BigDye Terminator Cycle Sequencing v3.1 kit and an ABI3100 genetic analyzer. i LUX primers without the hairpin structure and the fluorophore were used in the double sequencing reaction. The origin/homology of the sequence was identified by using the BLAST program. 2
In order to determine the sensitivity of the LUX RT-PCR assay as referred to the virus titer, serial 10-fold dilutions of chorioallantoic fluid containing the LaSota NDV vaccine strain as template in the range of 10+9.2 to 10+1.2 EID50/0.2 ml was tested by the assay. Each reaction was carried out in triplicate. The endpoint of detection was also determined for spiked chicken fecal samples using the same reaction conditions (virus strain, dilution steps). Besides the biological titers, the limit of detection was expressed in plasmid copy number by using the 10-fold dilution series of a TOPO Cloning Vector a containing the whole F gene of NDV strain KR-5/98 (courtesy of Dr. B. Lomniczi) as a standard target for the amplification. The copy number of the plasmid was calculated based on its molecular mass. The quantification was performed over a range of 2 × 1011 to 2 × 100 starting plasmid copy numbers.
Experimental infections of chickens and spiking of chicken feces were conducted in order to test the LUX RT-PCR assay on clinical specimens. In the animal experiments, tracheal and cloacal swabs and 10 different organ samples from brain, kidney, and lungs were collected from 4-week-old chickens experimentally infected with the LaSota strain of NDV (10 6 EID50/ml) 2 days postinfection. The specimens were subjected to LUX RT-PCR and virus isolation as described below. The spiking experiment was carried out by using 1 g of chicken feces samples that were previously proven free of NDV by egg inoculation and RT-PCR. These samples were spiked with 1 ml of 10-fold dilutions of NDV LaSota stock solution (original titer: 10+9.2 EID50/0.2 ml, final dilution: 10 9 ). Spiked samples were suspensed in distilled water and these 10% (wt/vol) suspensions were processed for RT-PCR as described below. The effects of possible residual inhibitory compounds in the fecal samples were tested by comparing the efficiency of the LUX RT-PCR runs using serial dilutions of purified RNA as target originating either from NDV-spiked feces and allantoic fluid of NDV-inoculated embryonated eggs.
The designed primer pair, the unlabeled member of which was degenerate, showed 100% of homology in most of the cases to the corresponding nucleotide sequences of archived and recent NDV isolates of various geographic origins available in GenBank (data not shown). All tested NDV strains were detected by the LUX assay, while no positive signal was obtained when heterologous pathogens, including the closely related avian paramyxovirus serotype 2 strains, were tested (Table 2). In the melting point analysis of the amplified products, the maximum peak of the curves was found consistently at 85.5°C ± 0.5°C. The nucleotide sequence of a representative PCR product was determined (GenBank acc. no.: DQ780002) and a BLAST search verified that a 143-nucleotide-long region of the NDV fusion protein gene was amplified in the reaction.
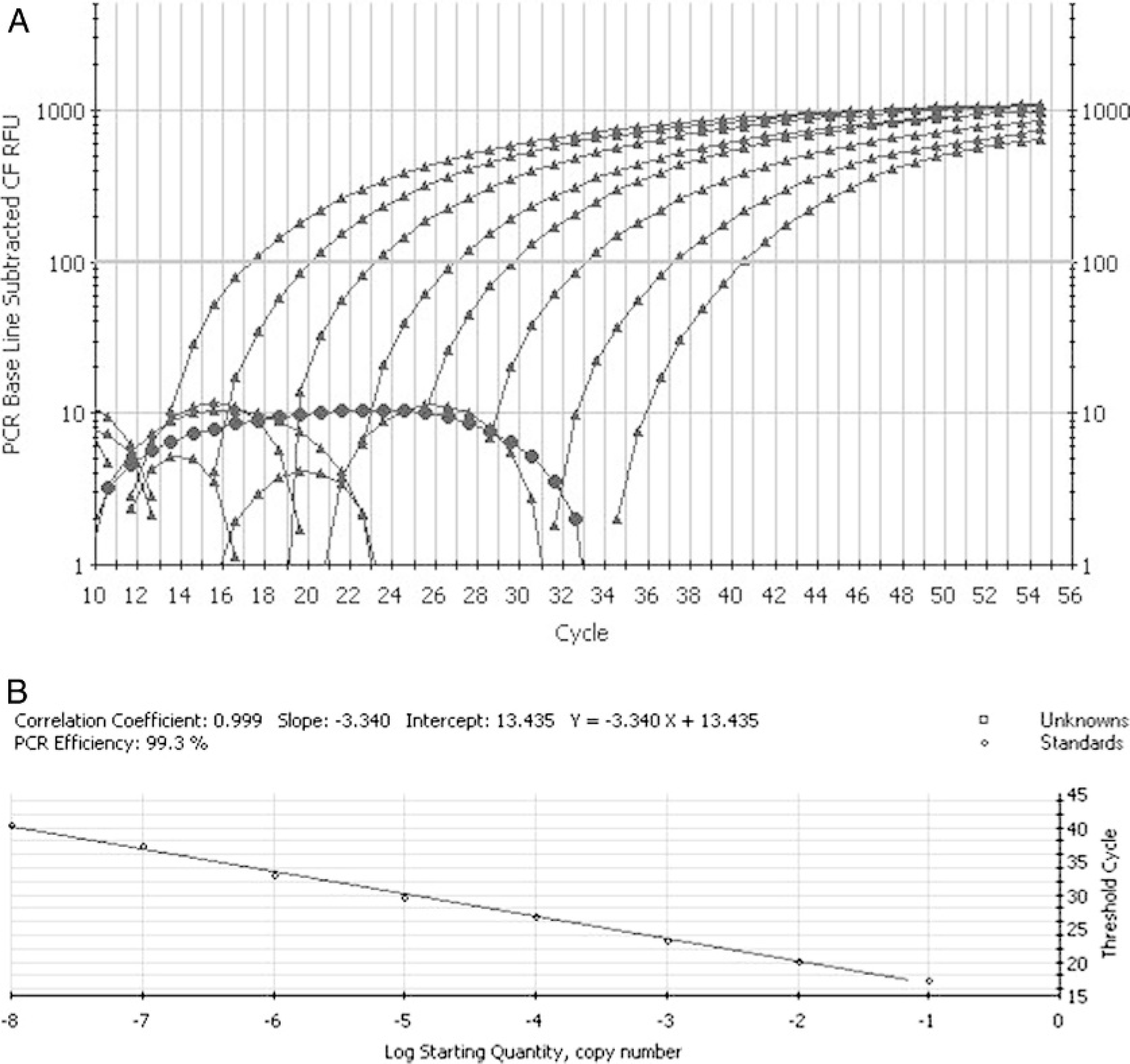
The analytical sensitivity of the assay proved to be 10+1.2 EID50/0.2 ml and 10+2.2 EID50/0.2 ml when tested on serial 10-fold dilutions of virus suspension and spiked chicken fecal samples, respectively. The standard curves generated from the amplification plots showed a linear correlation between CT values and the virus dilutions (Fig. 1). The assay was able to detect approximately 20 copies of the plasmid that contained the target gene (data not shown). The lower analytical sensitivity in fecal samples may be attributed to compounds such as bile salts, hemoglobin derivatives that are known to exert inhibitory effect on the performance of PCR. 6 However, the RNA dilution experiments revealed no residual inhibitory effects in the purified samples, therefore the sensitivity differencies should be due to the degradation of the RNA and/or the lower efficiency of viral RNA extraction from feces. Ten of each were investigated: the same 8 tracheal swabs, 4 cloacal swabs, 6 eyelid, and 6 lung specimens produced positive results when samples originating from experimentally infected chickens were investigated by either the LUX RT-PCR and virus isolation. The results of the assay (i.e., the dynamics and the endpoint of detection) were the same on the tested different real-time PCR instruments (data not shown).
Footnotes
a.
Invitrogen, Carlsbad, CA.
b.
Ceva-Phylaxia, Budapest, Hungary.
c.
Scientific and Educational Software, State Line, PA.
d.
Qiagen, Hilden, Germany.
e.
Amersham Life Science, Budapest, Hungary.
f.
Bio-Rad, Hercules, CA.
g.
Applied Biosystems, Foster City, CA.
h.
Corbett Research, Mortlake, NSW, Australia.
i.
Biomi, Göudöullöu, Hungary.