
Abstract
The usefulness of reverse transcription-polymerase chain reaction (RT-PCR) from formalin-fixed, paraffin-embedded (FFPE) tissues was examined and compared to the immunohistochemistry (IHC) and in situ hybridization (ISH) assays for detection of Newcastle disease virus (NDV). Spleen and lung tissues were collected from chickens experimentally infected with either of 2 NDV isolates: a low virulent virus (LaSota) and a virulent virus (from the 2002–2003 California outbreak). The tissues were harvested immediately postmortem and fixed in 10% neutral buffered formalin for approximately 52 hours. Also, just before euthanasia, oral and cloacal swabs were collected for virus isolation. RNA was obtained from the FFPE tissues by digestion with proteinase K and subsequent extraction with phenol, chloroform, and isoamyl alcohol. By seminested RT-PCR with primers for the NDV matrix gene, a 232-base pair (bp) product was generated and visualized by electrophoresis. The results of PCR were compared to those of IHC for viral nucleoprotein and ISH for matrix gene (850 bp) on 3-μm sections and to those of virus isolation from swabs. All samples from infected chickens were positive by RT-PCR, including samples that were negative by both IHC and ISH. The RT-PCR positives included tissue from chickens that were no longer shedding virus detectable by virus isolation. The RT-PCR was an effective and sensitive method to detect NDV in FFPE tissues. To the authorsn' knowledge, this is the first report of NDV detection in FFPE tissues as a diagnostic approach possibly suitable for archival materials.
Newcastle disease virus (NDV), a synonym for avian paramyxovirus type 1, is a nonsegmented, single-stranded, negative-sense, enveloped RNA virus 2 composed of approximately 15,200 nucleotides. 11 The clinical signs and gross and microscopic lesions of NDV infections are varied and are dependent on many factors related to the host, virus strain, and environmental stress, and there are no diagnostic lesions. 2 The use of immunohistochemistry (IHC) 7,8 and in situ hybridization (ISH) 3,4,7,8 with formalin-fixed, paraffin-embedded (FFPE) tissues has been valuable for determining virus distribution in samples from pathogenesis studies of NDV and would be of similar value for diagnostic applications. Reverse transcription-polymerase chain reaction (RT-PCR) has also been used for NDV detection in fresh/frozen tissues, 10 amnioallantoic fluid of infected embryonating eggs 10,12 and feces, 5 and for preparing cDNA from FFPE tissues for sequence analysis. 6
Formalin-fixed tissues can be handled safely in any laboratory, whereas handling of the infective samples of virulent NDV strains is restricted to biosecure conditions in the United States, defined as Biosafety Level 3 Agriculture (BSL-3Ag). In this study, therefore, a seminested RT-PCR method was developed to detect the NDV matrix gene from FFPE tissues. To show the sensitivity of the RT-PCR, the results were compared to those of IHC for viral nucleo-protein and ISH for matrix gene on spleen and lung FFPE tissues from NDV-infected and noninfected birds. The RT-PCR data were compared to results of virus isolation from oral and cloacal swabs collected from the sampled birds just before euthanasia.
Formalin-fixed, paraffin-embedded spleen and lung tissues from 4-week-old specific pathogen free (SPF) White Leghorn chickens inoculated conjunctivally with 1 of 2 NDV isolates, LaSota (low virulent) 16 and CA/S0212679 (from the 2002–2003 outbreak in California, known as CA END virus; virulent), 14 were used for RT-PCR, IHC, and ISH. For negative controls, tissues from chickens similarly inoculated with phosphate buffered saline were used. Immediately before euthanasia, oral and cloacal swabs were obtained from each bird for virus isolation. Sampled tissues were collected at different times postinoculation and fixed by immersion in 10% neutral buffered formalin for approximately 52 hours and were routinely processed into paraffin. For IHC and ISH, 3-μm sections were cut from the paraffin blocks. 3 A comparison of the 3 assays was completed on one of the chickens sampled each day at 5 and 10 days postinoculation (dpi) for the low virulent strain (LaSota) and at 2, 3, and 5 dpi for the virulent strain (CA END virus).
Tissues were examined by IHC with the following protocol to detect viral nucleoprotein. 3,8 After deparaffinization, tissue sections were subjected to antigen retrieval by microwaving for 10 minutes at full power in Vector antigen unmasking solution a followed by blocking with universal blocking reagent b as recommended by the manufacturer. The primary antibody, made in rabbit, was anti-peptide (nucleoprotein), used at 1:8,000 dilution. 7 The detection system was avidin-biotin-alkaline phosphatase a and the substrate was Vector Red. a The stained sections were counterstained lightly with hematoxylin and coverslipped. 15
Tissue sections for ISH were probed with a negative sense digoxigenin-labeled 850-base riboprobe representing the 5′ end of the matrix gene of B1 and Fontana for LaSota and CA END virus, respectively. The protocol was as described previously. 4,7,8 Briefly, tissue sections were deparaffinized, rehydrated, and digested with 30 μg/ml proteinase K for 15 minutes at 37°C. Hybridization was conducted overnight at 42°C with approximately 20 ng of probe in a prehybridization solution. After stringent washes, antidigoxigenin alkaline phosphate was added to the sections. The development was with chromogen/substrate nitroblue tetrazolium c and 5-bromo,4-chloro,3-indolylphosphate. c Tissues were counterstained lightly with hematoxylin and coverslipped. 3
The RNA extraction protocol was modified from a previously described method for canine distemper virus. 13 Each formalin-fixed tissue embedded in paraffin blocks was trimmed with a razor to obtain 35 mg of tissue, which was then placed into a microcentrifuge tube. These tissue pieces were then deparaffinized with 1 ml of deparaffinizing agent, d mixed on a vortex mixer, and centrifuged at 10,000 × g at room temperature for 10 minutes. The supernatant was discarded, and the deparaffinization step was repeated. The resultant tissue pellet was washed with 1 ml of cold 100% ethanol and dried for 15 minutes in a vacuum centrifuge. For digestion of proteins and nucleases, the dried sample was incubated at 50°C for 4 hours with 600 μl of digestion buffer (20 mM Tris-HCl pH 7.6, 20 mM ethylenediaminetetraacetic acid, and 1% sodium dodecyl sulfate with 300 μg of proteinase-K). To extract RNA, 600 μl of phenol:chloroform 5:1 was added to each tube, and the tubes were centrifuged at 10,000 × g at 4°C for 3 minutes. This step was repeated with the top layers. Then, 600 μl of chloroform:isoamyl alcohol 24:1 was added to the top layers collected, and the resulting mixture was centrifuged at 10,000 × g at 4°C for 3 minutes. For precipitation of purified nucleic acid, the top sample layers were mixed with 50 μl of sodium acetate (pH 7.0) and 1 ml of cold 100% ethanol and placed at −80°C overnight. The precipitated nucleic acid was centrifuged at 14,000 × g for 30 minutes at 4°C, and the pellet was washed with cold 70% ethanol and dried in a vacuum centrifuge for 15 minutes. The pellet was resuspended in 30 μl of nuclease-free water and stored at −80°C. All buffers and reagents were prepared with ribonuclease-free water. Total nucleic acid yielded was calculated by absorbance at 260 nm with a spectrophotometer. e For each RT-PCR, 1 μg nucleic acid was used.
The primers used for RT-PCR were generated by alignment of published NDV nucleotide sequences, representing part of the open reading frame for the matrix gene. The sequences of the outer set of primers were as follows: M0 (sense), 5′-GAGTTACTTTCYKCTGCRATGCTCTGCCTAGG-3′ (designed for this study); M2 (antisense), 5′-GTCCGAGCACATTGAGC-3′ 12 (574 base pair [bp]). The seminested primers were M1 (sense), 5′ -TCGAGICTGTAIAATCTTGC-3′ 12 and M2 (antisense) (232 bp). These primer sequences are known to be useful in the characterization of a large number of NDV isolates. 12 For reverse transcription, 1 μg of extracted nucleic acid was added to the following reaction mix: 1 μl of oligo (dT) f 12-18 (500 μg/ml), 1 μl of 10 mM dNTP mix, f and nuclease-free water to a total reaction volume of 17 μl. As positive control, 1 μl of NDV nucleic acid collected from amnioallantoic fluid of an embryonating egg infected with LaSota. For a negative control, either the extract from noninfected tissues or nuclease-free water was added instead of sample nucleic acid. The reaction was incubated in a 65°C bath for 5 minutes and then placed on ice for 2 minutes. The contents were collected by brief centrifugation and mixed with 4 μl of 5 × first-strand buffer, g 2 μl of 0.1 M dithiothreitol (DTT), g 1 μl of RNaseOUT Recombinant Ribonuclease, g and 1 μl of Superscript II RNase H- Reverse Transcriptase. g The reaction was incubated in a 50°C bath for 1 hour and then inactivated by heating at 70°C for 15 minutes.
The PCR was performed in a 50-μl volume containing 3 μl of sample cDNA, 25 μl of TaqPCR Master Mix, h 0.5 μl of each primer (M0 and M2), and 21 μl of nuclease-free water. The reaction was then incubated in a thermocycler i under the following conditions: 94°C for 5 minutes; 35 cycles of 94°C for 30 seconds (denaturation), 55°C for 30 seconds (annealing), and 72°C for 1 minute (extension). The samples were then chilled at 4°C. For the second round of PCR, 3 μl of the first PCR product was amplified by using the same reaction conditions used for the first PCR, with the primers M1 and M2.
A total of 5 μl of each seminested PCR product was subjected to agarose gel electrophoresis on a 2.0% agarose gel in TAE buffer (40- mM Tris-acetate, 2-mM ethylene-diaminetetraacetic acid [EDTA]) for 45 minutes at 90 V. For staining, 1.25 μl of ethidium bromide per 30 ml was added to the gel. The gel was photographed with UV illumination. For determination of the size of the PCR products, an ethidium bromide-stainable 100-bp DNA ladder f was subjected to gel electrophoresis as a marker. A band of the spleen sample infected 10 days previously with LaSota was excised from the gel, and nucleic acids were extracted. j The extracted DNA fragments were sequenced (Molecular Genetics Instrumentation Facility, University of Georgia).
Oral and cloacal swabs for virus isolation were obtained from each bird and placed in a tube containing 1.5 ml of brain heart infusion broth with antibiotics (2,000 unit/ml penicillin G, 200 μg/ml gentamicin surface, and 4 μg/ml amphotericin B k ). Swab fluids were centrifuged at 1,000 × g for 20 minutes, and the supernatant was inoculated into 9- to 10-day-old SPF embryonating chicken eggs. Newcastle disease virus-infected dead or surviving embryos were identified by hemagglutination (HA) activity in allantoamniotic fluid harvested from chilled eggs. Newcastle disease virus was confirmed in HA-positive samples by hemagglutination-inhibition test with NDV-specific antiserum. 2
Viral infection detected by IHC, ISH, and RT-PCR in spleen and lung from formalin-fixed, paraffin-embedded tissues and virus isolation of oral and cloacal swab samples from a Newcastle disease virus-infected chicken sampled each day.
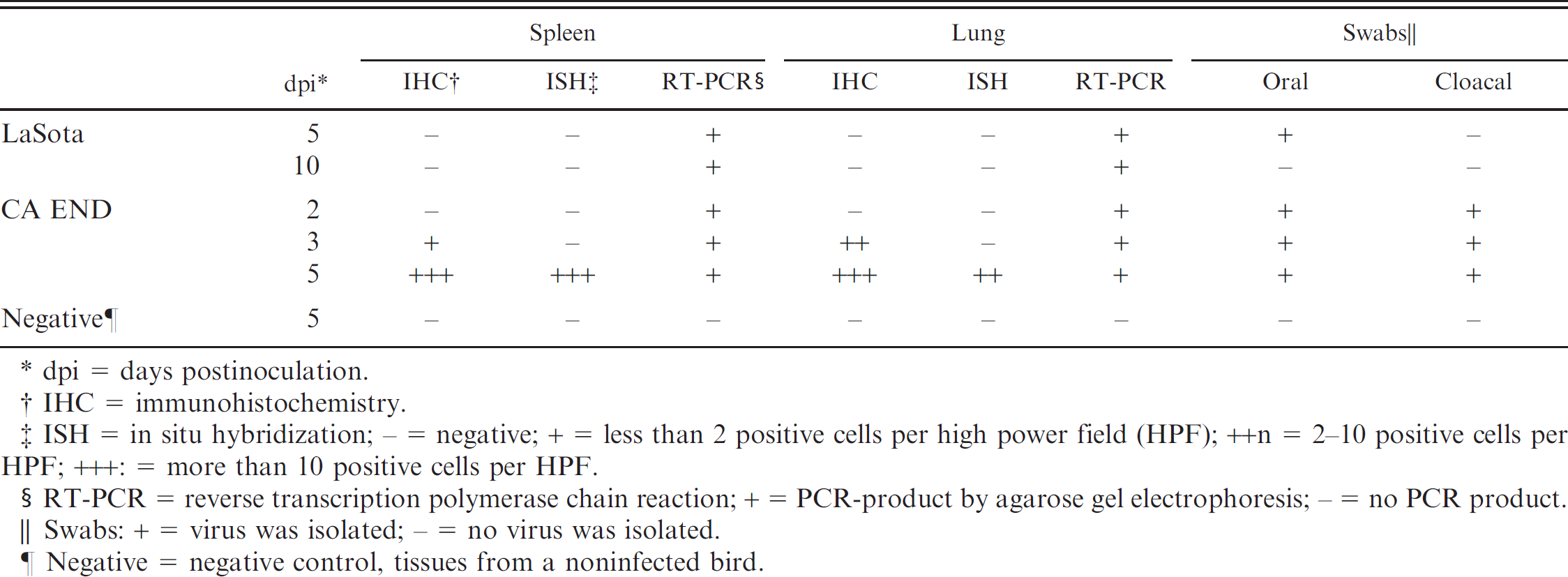
dpi = days postinoculation.
IHC = immunohistochemistry.
ISH = in situ hybridization; - = negative; + = less than 2 positive cells per high power field (HPF); ++n = 2–10 positive cells per HPF; +++: = more than 10 positive cells per HPF.
RT-PCR = reverse transcription polymerase chain reaction; + = PCR-product by agarose gel electrophoresis; - = no PCR product.
Swabs: + = virus was isolated; - = no virus was isolated.
Negative = negative control, tissues from a noninfected bird.
Results of IHC, ISH, and RT-PCR on FFPE tissues and virus isolation of swabs are presented in Table 1. A specific NDV product, a part of matrix gene (232 bp), was amplified from all examined tissues from birds infected with either LaSota or CA END virus at the sampled time points. Figure 1 illustrates results of the seminested RT-PCR amplifications from spleen tissue. Results from lung tissue were similar to those from spleen tissue shown in Figure 1. The nucleotide sequence analysis of the PCR product from the spleen sample collected from a bird at 10 dpi with LaSota demonstrated more than 97% identity with the published LaSota matrix gene nucleotide sequence (GenBank U25833). The result confirmed that the PCR product was consistent with that of the NDV matrix gene.
Newcastle disease virus antigen was detected by IHC in both spleen and lung tissues with CA END virus at 3 and 5 dpi. In the spleen, scattered macrophages and lymphocytes were IHC-positive at 3 dpi, and the numbers of positive cells were increased at 5 dpi. In the lung, IHC-positive macrophages and lymphocytes were limited to within the bronchus associated lymphoid tissue (BALT) at 3 dpi, but positive cells extended into the interstitium at 5 dpi. NDV antigen was not detected by IHC in both the spleen and lung with LaSota at 5 and 10 dpi and with CA END virus at 2 dpi. In situ hybridization was positive in both the spleen and lung only with CA END virus and only at 5 dpi. Scattered macrophages and lymphocytes within the spleen were ISH positive. In the lungs with CA END virus, hybridization occurred within macrophages and lymphocytes in the interstitium as well as in BALT. Virus isolation detected virus shedding to confirm the birds were infected and not whether the sampled tissues were actually virus positive. In birds infected with LaSota, only the oral swab sample collected at 5 dpi yielded virus. In contrast, virus was isolated from both oral and cloacal swabs of birds infected with CA END virus at all time points examined. No virus was detected in spleen and lung tissues or swabs from noninfected birds.
In summary, all 10 samples from infected birds were positive by RT-PCR of FFPE tissues, and FFPE tissues from noninfected birds were negative. In comparison, IHC-positives were detected in only 4 of 10, and ISH-positives were detected in only 2 of 10 of those tissue blocks. These results indicated that the RT-PCR with FFPE tissues is an effective and sensitive method to detect NDV.
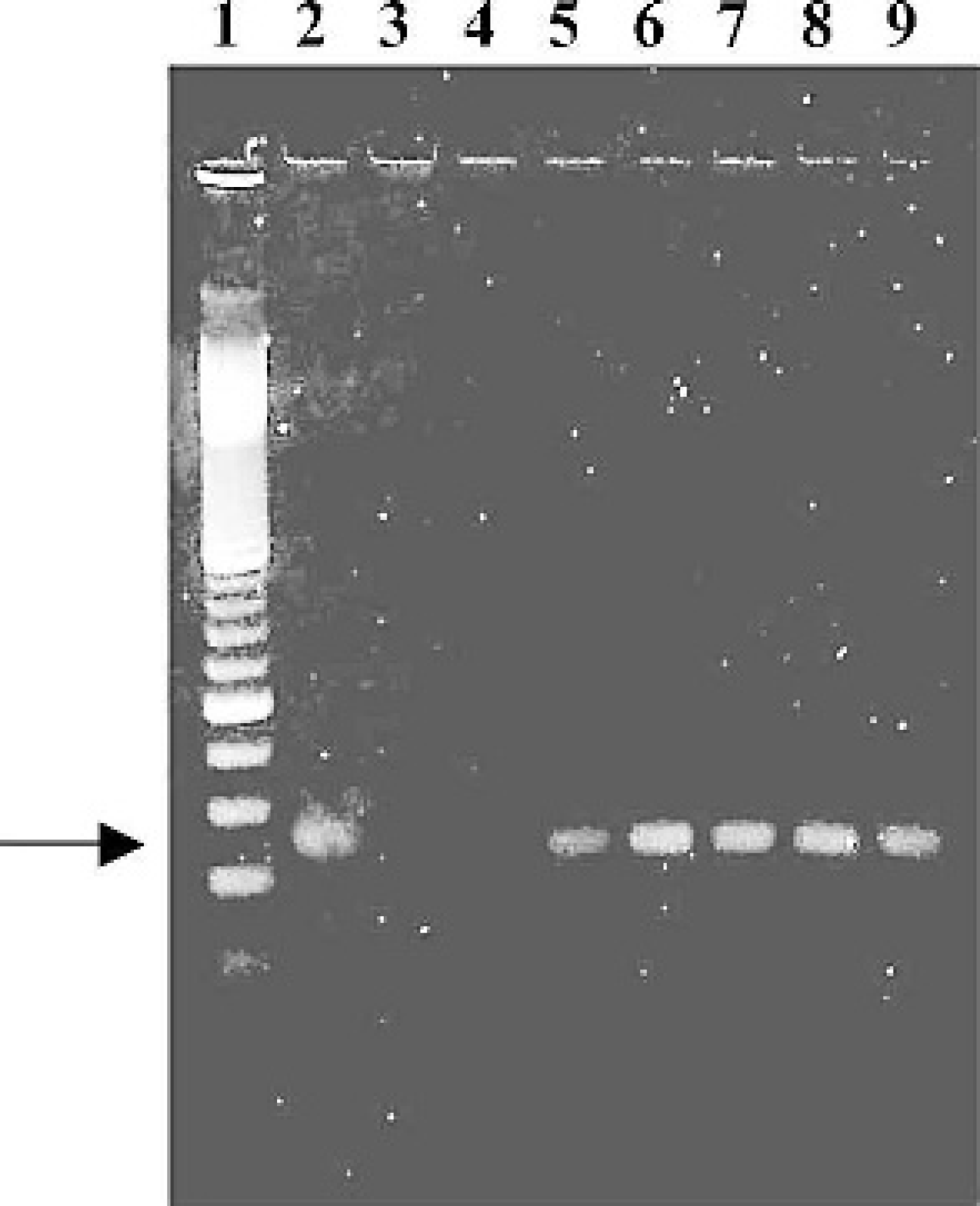
Agarose gel (2%) electrophoresis of seminested RT-PCR products from formalin-fixed, paraffin-embedded Newcastle disease virus-infected chicken spleen tissue. Lane 1, 100-bp step ladder; lane 2, positive control; lane 3, water-only negative control; lane 4, noninfected negative control; lane 5, LaSota 5 dpi; lane 6, LaSota 10 dpi; lane 7, CA END virus 2 dpi; lane 8, CA END virus 3 dpi; lane 8, CA END virus 5 dpi. Arrow, band of expected 232-bp product from seminested RT-PCR in lanes 2, 5, 6, 7, 8, and 9.
This RT-PCR technique could be used to detect infection from archival or fixed materials, for virulent as well as low virulent viruses. As an auxiliary advantage of RT-PCR, it is possible to sequence the PCR product, enabling more extensive analysis of changes in the genome and for molecular epidemiologic studies. In this experiment, the matrix gene was chosen for amplification, but the application of primers for detection of the fusion gene could yield valuable information regarding potential virulence changes. 1,12 The success of the development of this RT-PCR from FFPE tissues enables work on NDV in more flexible conditions because FFPE tissues can be handled without biosecurity concerns.
However, the RT-PCR has several disadvantages. Ideally, tissues should be fixed rapidly postmortem, as enzymes active during autolysis will destroy RNA, which is relatively labile. This is not always possible under field conditions. Also, after fixation, subsequent processing into paraffin should not be delayed because prolonged fixation of tissues can severely limit their usefulness as formalin can break nucleic acid strands. The size of the desired products should be small because of an increase of the percentage of false-negative results as the size of the amplicon increases. 9,17 In this study, RT-PCR was positive in the spleen tissues from chickens infected with LaSota. In general, with low virulent strains there is a limited distribution of virus to systemic organs, in contrast to virulent strains that are widely disseminated. Although there were positive RT-PCR results on the spleens from chickens infected with LaSota, there is no information on cell types or the number of cells affected, because RT-PCR does not provide that type of information. By comparison, the IHC and ISH provide valuable information by demonstrating the distribution of viral nucleoprotein and mRNA of matrix gene, respectively, within tissues and can highlight the types and numbers of positive cells. Therefore, IHC/ISH in addition to the RT-PCR from the FFPE tissues can complement the results of RT-PCR, which may be a more sensitive method than IHC/ISH.
In conclusion, we successfully developed a method to detect NDV in FFPE tissues by RT-PCR and demonstrated its use as potential diagnostic tool as well as a technique for future use in NDV research. Furthermore, it is possible to perform retrospective studies of archived materials from cases where ND was not the initial diagnosis.
Acknowledgements. We acknowledge the excellent technical support of Jian Zhang, Dr. James Stanton, Cory Gresham, and Taylor Deal. This work was supported by U.S. Poultry & Egg Association (project number 368) and the USDA Agricultural Research Service (CRIS number 6612-32000-038-OOD). Mention of trade names or commercial products in this article is solely for the purpose of providing specific information and does not imply recommendation or endorsement by the U.S. Department of Agriculture.
Footnotes
a.
Vector Laboratories, Burlingame, CA.
b.
Biogenex, San Ramon, CA.
c.
Roche, Indianapolis, IN.
d.
Citrisol, Fisher Science, Pittsburgh, PA.
e.
Ultrospec 3000, Pharmacia Biotech, Piscataway, NJ.
f.
Promega, Madison, WI.
g.
g. Invitrogen, Carlsbad, CA.
h.
Qiagen, Valencia, CA.
i.
PCR Sprint Thermal Cycler, Bioscience Technologies, Milford, MA.
j.
QiaQuick Gel Extraction kit, Qiagen, Valencia, CA.
k.
Sigma Chemical Co., St. Louis, MO.