
Abstract
Molecular classification of colorectal cancer is difficult to implement in clinical settings where hundreds of genes are involved, and resources are limited. This study aims to characterize the molecular subtypes of patients with sporadic colorectal cancer based on the three main carcinogenic pathways microsatellite instability (MSI), CpG island methylator phenotype (CIMP), and chromosomal instability (CIN) in a Chilean population. Although several reports have characterized colorectal cancer, most do not represent Latin-American populations. Our study includes 103 colorectal cancer patients who underwent surgery, without neoadjuvant treatment, in a private hospital between 2008 and 2017. MSI, CIN, and CIMP status were assessed. Frequent mutations in KRAS, BRAF, and PIK3CA genes were analyzed by Sanger sequencing, and statistical analysis was performed by Fisher’s exact and/or chi-square test. Survival curves were estimated with Kaplan–Meier and log-rank test. Based on our observations, we can classify the tumors in four subgroups, Group 1: MSI-high tumors (15%) are located in the right colon, occur at older age, and 60% show a BRAF mutation; Group 2: CIN-high tumors (38%) are in the left colon, and 26% have KRAS mutations. Group 3: [MSI/CIN/CIMP]-low/negative tumors (30%) are left-sided, and 39% have KRAS mutations; Group 4: CIMP-high tumors (15%) were more frequent in men and left side colon, with 27% KRAS and 7% presented BRAF mutations. Three percent of patients could not be classified. We found that CIMP-high was associated with a worse prognosis, both in MSI-high and MSI stable patients (p = 0.0452). Group 3 (Low/negative tumors) tend to have better overall survival compared with MSI-high, CIMP-high, and CIN-high tumors. This study contributes to understanding the heterogeneity of tumors in the Chilean population being one of the few characterizations performed in Latin-America. Given the limited resources of these countries, these results allow to improve molecular characterization in Latin-American colorectal cancer populations and confirm the possibility of using the three main carcinogenic pathways to define therapeutic strategies.
Introduction
Colorectal cancer (CRC) is a heterogeneous disease of high relevance, being the third cancer-related cause of death in men and second in women worldwide. 1 In Chile, CRC mortality has significantly increased in recent years, 2 becoming the third cancer-related cause of death in the country. 3 The carcinogenesis of sporadic CRC results from the sequential accumulation of genetic and epigenetic alterations, and its main molecular pathways include chromosomal instability (CIN), microsatellite instability (MSI), and CpG island methylator phenotype (CIMP).4,5 According to the molecular classification of CRC, four consensus molecular subtypes (CMS) were established related to clinical, pathological, and biological characteristics. This current CMS classifier is based on a signature involving hundreds of genes,5–7 which is difficult to implement in clinical settings where resources could be limited. Approximately 15% of sporadic CRC patients have MSI-high (MSI-H) tumors, which confers good prognosis at an early stage or good response to immunotherapy at an advanced stage, but most patients are MSI stable (MSS) and show poor response to immunotherapy.8,9 MSI-H compared with MSS tumors have highly upregulated expression of various immunological checkpoints, which are being investigated in clinical trials as possible therapeutic targets (PD1, PDL1, or CTLA4).10,11 Furthermore, BRAF mutations are diagnosed in 5%–15% of CRC patients, being a known biomarker related to a less favorable outcome. 12 Sporadic MSI-H CRC is associated with a BRAF mutation in 50% of cases. This association varies depending on the tumor stage; there are reports suggesting a higher frequency of mutated BRAF in MSI-H/metastatic CRC (34.6%) compared with early-stage MSI-H tumors (24%), being MSI-H/BRAFmut a more aggressive phenotype. 12 While several reports show differences regarding the survival of patients according to their molecular status, the role of MSI as a prognostic factor remains controversial. 13
Likewise, approximately 30%–40% of sporadic proximal CRCs are CIMP-high (CIMP-H), compared with 3%–12% of distal CRCs. 14 CIMP-H patients have shown a worse prognosis. 15 It has been reported that between 25% and 60% of CIMP-H tumors have MSI-H due to errors introduced by the mismatch repair (MMR) system caused by hypermethylation in the MLH1 promoter. Although some studies have proposed CIMP-H as a predictive biomarker for personalized therapy in CRC patients, results have not been conclusive. 15 This could be explained by the high heterogeneity of CIMP-H tumors given by the presence/absence of MSI-H that confers a different immuno-environment to both subtypes. 16 Thus, the severity of the prognosis could vary in different subgroups of CRC depending on MSI, CIMP, KRAS, BRAF, and PIK3CA mutations’ status and tumor stage.17–23
Several reports have characterized CRC; however, most do not represent the Latin-America population. Given the limited resources of these countries to evaluate all genes involved in consensus classification, we propose to characterize Chilean CRC patients according to a sub-molecular classification based on the three principal carcinogenic pathways.
Materials and methods
Study design
Exploratory analytical study of 103 consecutive CRC patients undergoing surgery between 2008 and 2017 at Clínica Las Condes. Exclusion criteria were neoadjuvant previous therapy, coexistence of inflammatory bowel disease, and hereditary CRC syndromes. Each patient signed an informed consent approved by the Institutional Bioethics Committee according to the local institutional review board (IRB) regulations. Demographic and clinicopathological characteristics were recorded.
Pathological data
Surgical specimens were examined and diagnosed based on American College of Pathology Protocol (CAP), World Health Organization (WHO), and AJCC Staging Manual (8th Edition). Since cases are collected during a long period of time, all of them were reevaluated for consistency and accuracy by a certificated pathologist. Pathological data are detailed in Supplementary methods S1. Representative tissue sections analyzed, and details are shown in Figure S1.
DNA extraction
Normal genomic DNA was purified from peripheral venous blood according to Wizard Genomic DNA Purification Kit (Promega). Tumor DNA from sections of formalin-fixed and paraffin-embedded (FFPE) tissue was extracted by the QIAamp DNA FFPE Tissue extraction kit (QIAGEN) from areas with ≥80% of tumor cells. Tumor DNA integrity was verified by multiplex polymerase chain reaction (PCR). 24
MSI status analysis
MSI was determined by analyzing seven microsatellite markers from the NCI panel (Bat-25 / Bat-26 / Bat-40 / D2S123 / D3S1029 / D5S346 / D17S250). Each microsatellite marker was PCR amplified from tumor and normal DNA as control sample. 25 Tumors were classified as MSI-H (≥3 unstable markers) and MSI-low/negative (MSI-L/0 or MSS) (<3 unstable markers).
CIMP status analysis
CIMP was determined by MethyLight 26 using a 5 markers panel (CACNA1G / IGF2 / NEUROG1 / RUNX3 / SOCS1) 27 plus MLH1 promoter region c.-277 to -194 (NM_000249.3). Tumor DNA samples were modified by the EZ DNA Methylation-Gold kit (Zymo Research). ALU-C4 was used as a reference gene for bisulfite conversion. Methylated DNA with Ssi I methyltransferase (Promega) was used as a positive control for methylation. Methylation is represented as a percentage relative to Ssi I treated DNA reference sample; a gene was considered methylated when over 80% methylation occurred. Tumors were classified as CIMP-high (CIMP-H) (≥ 3 methylated genes) and CIMP-low/negative (CIMP-L/0) (< 3 methylated genes).
CIN status analysis
Deletion analysis of 10 short tandem repeats (STRs) adjacent to APC (D5S134-D5S346-D5S656-D5S82), DCC (D18S46-D18S64-D18S69) and TP53 (D17S1176-D17S1881-D17S250) genes was performed by PCR 28 from tumor and normal DNA. Loss of heterozygosity (LOH) was positive when the relative peak height of tumor DNA versus normal DNA decreases by 30%. Tumors were classified as CIN-high (CIN-H) ≥ 3 STRs with LOH and CIN-low/negative (CIN-L/0) < 3. 29
Somatic mutations in KRAS, BRAF, and PIK3CA analysis
Mutations in KRAS (codons 12 and 13), BRAF (codon 600), and PIK3CA (exons 9 and 20) genes were amplified by PCR from tumor DNA 30 and detected by Sanger sequence.
Statistical analysis
Categorical variables were described with absolute and percentage frequency, continuous variables, with median and minimum–maximum interval. To compare categorical variables, Fisher’s exact and/or chi-square tests were performed; overall survival was determined from the endpoint defined as the time from surgery until the date of death due to illness or censored on the date of last clinical follow-up; no patients were discarded due to lack of follow-up. The effect of different variables on survival was evaluated with univariate Cox proportional hazard analysis, Kaplan–Meier survival curves, and differences between groups by log-rank test. The statistical software SPSS v24 IBM was used.
Results
Tumors from 103 sporadic CRC patients were analyzed and a clinicopathological database was generated from data collected from patient’s clinical record and pathology report. These data have been summarized in Table 1. The median age of these patients was 66 years (range: 38−97) and 65% (67/103) were male. Male patients had tumors more frequently located in the left colon (76% vs 53%, p = 0.026), with a higher age (≥60 years) at diagnosis (67% vs 25%, p < 0.0001) and a higher body mass index (BMI) ≥ 25 kg/m2 (60% vs 36%, p = 0.015) compared with female patients. The average follow-up of this cohort was 56 months (range: 1–132).
Clinicopathological characteristics of patients with colorectal cancer.

Values in the table represent numbers followed in parenthesis by %. Asterisks mean that the p value is statistically significant. BMI: body mass index; NI: not informed; Mut: mutated; Wt: wild type; UnMeth: unmethylated; Meth: methylated; AJCC: American Joint Committee on Cancer; TNM: tumor−node−metastasis, TILs: tumor infiltrating lymphocytes.
Distribution of MSI and clinicopathological characteristics
In this cohort, MSI-L/0 (MSS) tumors were more frequent (88/103; 85%) than MSI-H tumors (15/103; 15%) (Table 2). A comparison between MSS versus MSI-H tumors showed that MSS tumors presented left side location more frequently 77% (68/88) versus 13% (2/15) (p < 0.0001), well/moderate differentiation 91% (80/88) versus 60% (9/15) (p < 0.0001), KRAS mutation 31% (27/88) versus 0% (p < 0.0001), CIN-H 55% (48/88) versus 0%, and unmethylated MLH1 97% (85/88) versus 33% (5/15) (p = 0.001), respect to MSI-H tumors. However, MSI-H tumors frequently exhibit hypermethylated phenotype (CIMP-H), 67% (10/15) versus 17% (15/88) p = 0.002, and BRAF mutation 60% (9/15) versus 3% (3/88) p = 0.001 as compared with MSS tumors.
Characteristics of patients with colorectal cancer classified by MSI status.
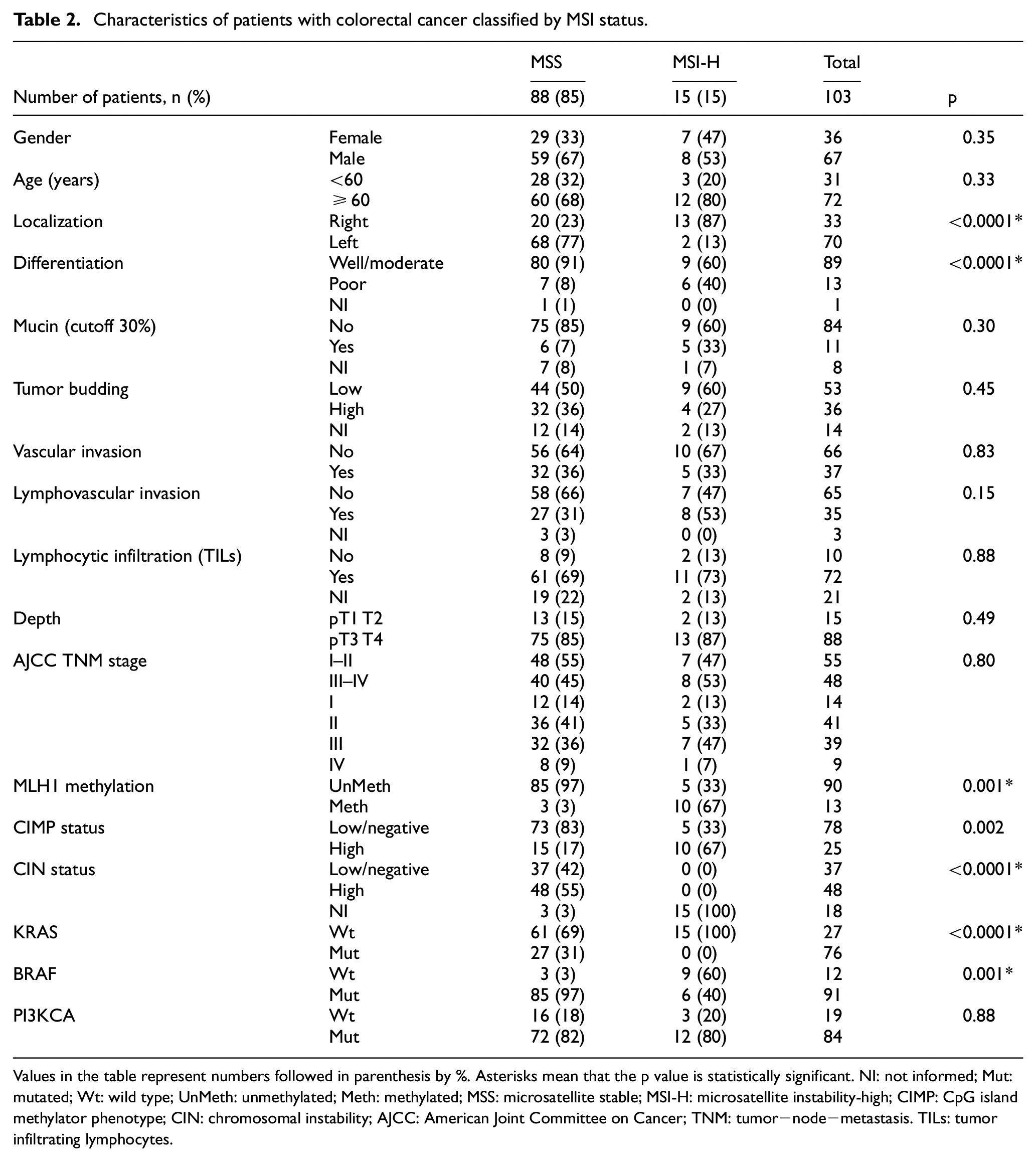
Values in the table represent numbers followed in parenthesis by %. Asterisks mean that the p value is statistically significant. NI: not informed; Mut: mutated; Wt: wild type; UnMeth: unmethylated; Meth: methylated; MSS: microsatellite stable; MSI-H: microsatellite instability-high; CIMP: CpG island methylator phenotype; CIN: chromosomal instability; AJCC: American Joint Committee on Cancer; TNM: tumor−node−metastasis. TILs: tumor infiltrating lymphocytes.
Distribution of MSI and mutations according to staging
MSI-H was observed in 13% (7/55) of stage I–II and 17% (8/48) of stage III–IV patients (Table 3). In early stages, there is a higher percentage of women (21%) with MSI-H than men (8%) p = 0.219, unlike late stages where men and women MSI-H frequency is similar (18% and 16%, respectively). In both early and late stages, MSI-H is observed in patients with right colon cancer, 36% (p = 0.009) and 42% (p < 0.0001), respectively. KRAS mutation was only observed in MSS patients, with a similar distribution between early (15/48) and late stages (12/40). In the early stages, BRAF mutations were only observed in patients with MSI-H, but in late stages 38% (3/8) (p < 0.002) of patients with mutated BRAF were MSS. For PIK3CA, 92% (11/12) of patients in the early stages were MSS, while in late stages a lower frequency with 71% (5/7) was observed. Regarding the CIMP-H phenotype in late stages, there is a higher number of patients with MSI-H (47%) than in early stages (30%). CIN was observed only in MSS patients without differences between early or late stages (Table 3).
Characteristics of patients with colorectal cancer classified by MSI status in early and late stages.

Values in the table represent numbers followed in parenthesis by %. Asterisks mean that the p value is statistically significant. NI: not informed; ND: not determinate; Mut: mutated; Wt: wild type; UnMeth: unmethylated; Meth: methylated; MSS: microsatellite stable; MSI-H: microsatellite instability-high; CIMP: CpG island methylator phenotype; CIN: chromosomal instability.
CIMP/CIN analysis according to clinicopathological characteristics
CIMP-H was observed in 24% (25/103) of tumors while 76% were CIMP-L/0. CIMP-H tumors are more frequently located on the right colon, have a mucinous component, and are poorly differentiated compared with CIMP-L/0 tumors (p < 0.005). In addition, CIMP-H presents 36% BRAFmut, 16% KRASmut, and 28% PIK3CAmut. MSI-H status was observed in 40% of CIMP-H tumors (Table S1). It was possible to analyze CIN status in 85 patients (82.5%) (Table S2), while 17.5% (18/103) were non-informative due to MSI or low tumor DNA concentration for LOH evaluation. Thus, 56% of analyzed patients were CIN-H, and 44% CIN-L/0. It was observed that 19% of CIN-H tumors have CIMP-H and all of them were MSS. Within this CIN-H group, tumors have 23% KRAS, 6% BRAF, and 17% PIK3CA mutations.
Relationship between MSI, CIMP, and mutational status of KRAS, BRAF, and PIK3CA genes
In 47% (48/103) of the analyzed tumors, at least one mutation was found in KRAS, BRAF, or PIK3CA genes (data not shown). MSS tumors present more mutations than MSI-H: 67% (32/48) in MSS/CIMP-L/0 tumors, 15% (7/48) in MSS/CIMP-H, 16% (8/48) in MSI-H/CIMP-H, 2% (1/48) in MSI-H/CIMP-L/0. The type and frequency of mutations in KRAS, BRAF, and PIK3CA genes according to MSI and CIMP status are shown in Figure 1. KRAS mutations are found only in MSS tumors regardless of CIMP status. Activating mutations on BRAF are preferentially found in MSI-H/CIMP-H tumors. BRAF and KRAS mutations mutually exclude each other. Noteworthy, patients with MSI-H/CIMP-0/L do not present mutations in PIK3CA (Figure 1). PIK3CA mutations were observed in 18% (19/103) of tumors, whereas concomitant mutations exist only in 10% (10/103) (data not shown).

Mutations percentages identified in colorectal cancer patients: MSS/CIMP-L/0: MSI stable/CIMP-low or stable; MSS/CIMP-H: MSI stable/CIMP-high; MSI-H/CIMP-L/0: MSI-high/CIMP-low or stable; MSI-H/CIMP-H: MSI-high/CIMP-high.
The analysis of carcinogenic pathways in our study population allowed us to identify four subgroups showing that 15% (15/103) of patients are MSI-H (Group 1), independently of the other pathways; 38% (39/103) are CIN-H (Group 2); and 30% (31/103) are low or stable [MSI/CIN/CIMP]-L/0 (Group 3). Regarding the methylator phenotype, 24% (25/103) are CIMP-H, 15% of those are CIMP-H with or without CIN-H and MSS (15/103) (Group 4), and 10% CIMP-H with MSI-H (10/103) that were included in Group 1 (Figure 2). Group 1 patients (MSI-H) show differences regarding (a) tumor localization, right versus left p < 0.0001; (b) stages of differentiation, poor versus well/moderate p = 0.008; (c) presence of mucin p = 0.007; (d) KRASwt p = 0.05, BRAFmut p < 0.0001; and (e) MLH1-methylated p < 0.0001 compared with CIN-H (Group 2), and [MSI/CIN/CIMP]-L/0 (Group 3) and CIMP-H (Group 4) (Table 4).
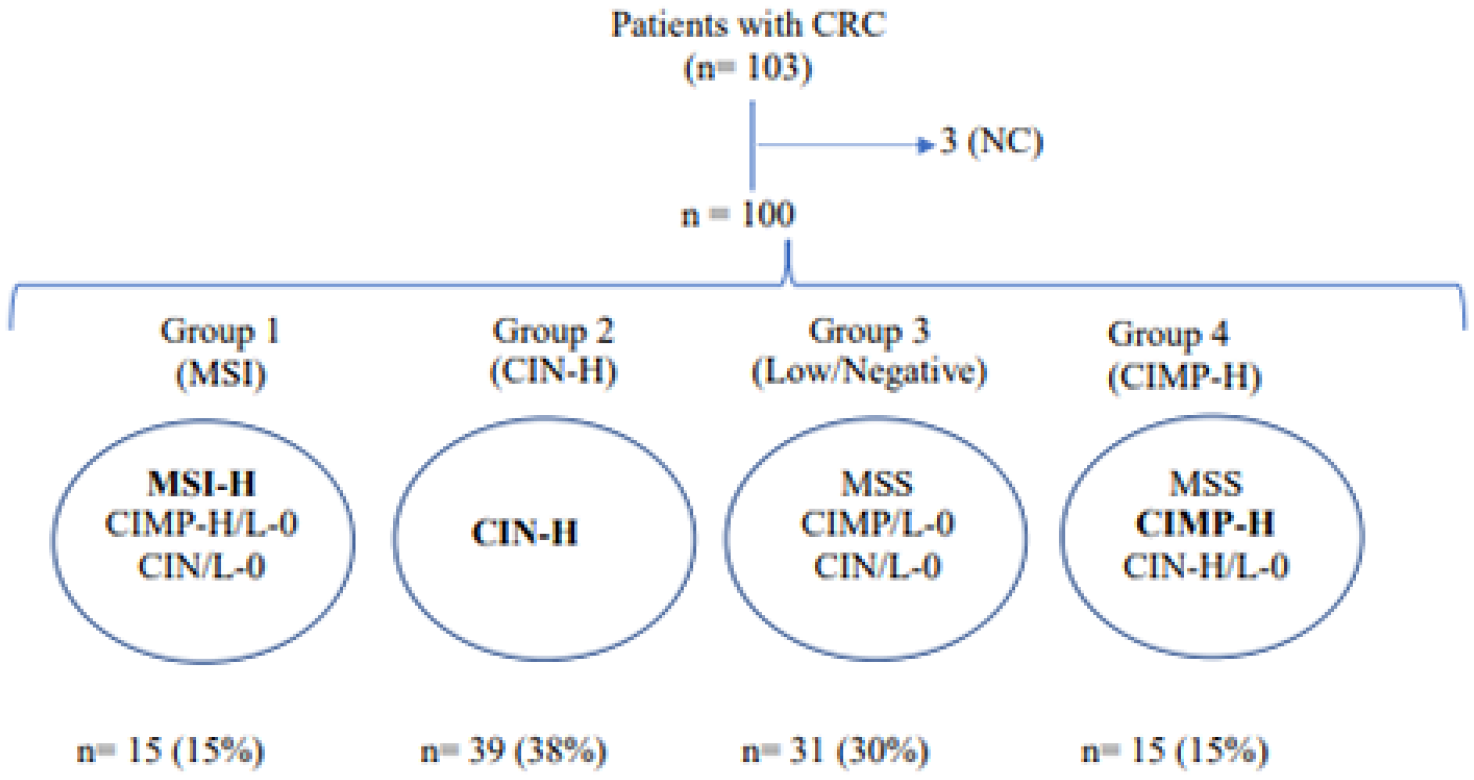
Flowchart of patients with colorectal cancer available for the analysis of the different carcinogenic pathways. CRC: colorectal cancer; CIN: chromosomal instability; MSI: microsatellite instability; CIMP: CpG island methylator phenotype; MSS: MSI stable; H: high; L: low; 0: negative; NC: non-classified.
Characteristics of patients with colorectal cancer classified into four groups: MSI-H, CIN-H, (MSI/CIN/CIMP)-low/0 and CIMP-H.
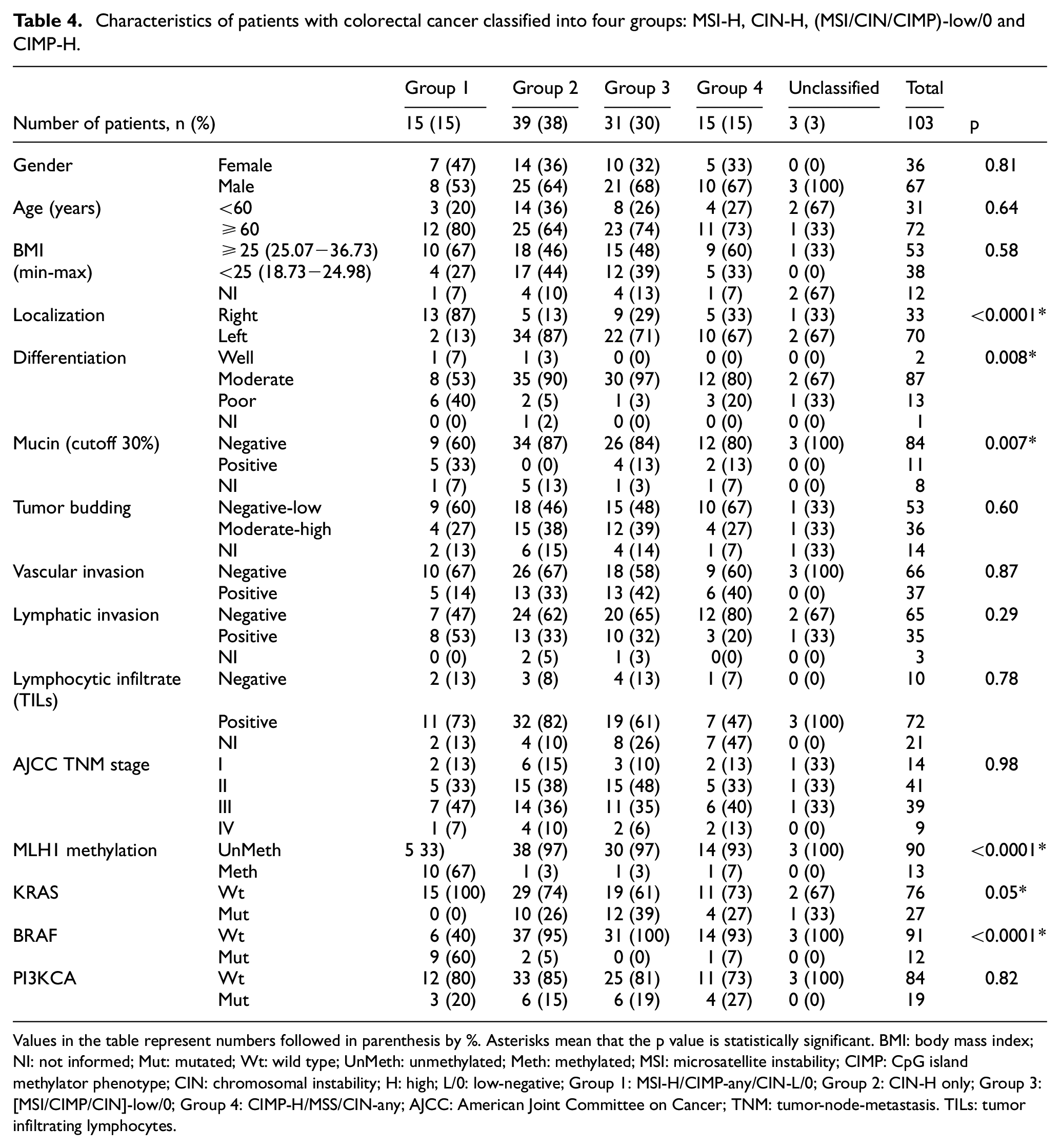
Values in the table represent numbers followed in parenthesis by %. Asterisks mean that the p value is statistically significant. BMI: body mass index; NI: not informed; Mut: mutated; Wt: wild type; UnMeth: unmethylated; Meth: methylated; MSI: microsatellite instability; CIMP: CpG island methylator phenotype; CIN: chromosomal instability; H: high; L/0: low-negative; Group 1: MSI-H/CIMP-any/CIN-L/0; Group 2: CIN-H only; Group 3: [MSI/CIMP/CIN]-low/0; Group 4: CIMP-H/MSS/CIN-any; AJCC: American Joint Committee on Cancer; TNM: tumor-node-metastasis. TILs: tumor infiltrating lymphocytes.
Patients’ survival association to MSI, CIMP, and mutations status
In this cohort, we did not observe a significant difference in overall survival (OS), between MSS and MSI-H patients, at either early or late stages (p = 0.2742) (Figure 3(a)). The CIMP pathway however showed that patients with CIMP-H tumors have worse prognosis compared with CIMP-L/0 at early or late stages (p = 0.0452) (Figure 3(b)).

Kaplan–Meier survival curves. (a): MSI-H versus MSS; (b): CIMP-H versus CIMP-0; (c): MSI/CIMP-0 versus MSI/CIMP-H; MSS/CIMP-0; MSS/CIMP-H.
Considering the conjoint effect of MSI and CIMP on survival, regardless the MSI status, patients with CIMP-H have worse but not statistically significant OS (p = 0.1952) (Figure 3(c)).
Concerning the subgroup classification, low/negative tumors (Group 3) have a slightly better OS (p = 0.5609) compared with MSI-H (Group 1), CIMP-H (Group 4), and CIN-H (Group 2) tumors (Figure 4(a)).

Kaplan–Meier survival curves. (a): MSI-H versus CIN-H; low/0; CIMP-H; (b): MSS/KRASmut versus MSS/PI3Kmut; MSS/KRAS-PI3Kmut; MSS/KRAS-PI3Kwt; (c) MSI/BRAFmut versus MSI/BRAF-PI3Kmut; MSI/BRAF-PI3Kwt.
To study the effect of mutations on the OS of MSS patients, KRAS and/or PIK3CA mutations were compared with wild type, excluding the three BRAF mutated patients. Under this aspect, patients with MSS/KRASmut have worse but not significant survival compared with MSS/PIK3CAmut, MSS/KRASmut/PIK3CAmut, and MSS/KRASwt/PIK3CAwt, p = 0.4140 (Figure 4(b)).
No KRAS mutations were observed in MSI-H patients. MSI-H/BRAFmut patients have worse survival compared with MSI-H/BRAFmut/PIK3CAmut and MSI-H/BRAFwt/PIK3CAwt (p = 0.1239) (Figure 4(c)). Differences between these groups at early or advanced stages could not be studied due to the small sample size.
Therefore, regardless of KRAS, BRAF, or PIK3CA mutational status in MSI-H or MSS patients, the worst survival was observed in CIMP-H patients. Furthermore, CIMP-H patients with mutated KRAS had a worse prognosis compared with KRASwt, BRAFwt, and BRAFmut (data not shown).
The univariate analysis for associations between clinicopathological factors and OS is shown in Table 5. Data from 103 patients were available for survival analyses. Across all samples, CIMP was significantly associated with OS (p = 0.03). Also, BRAF mutation (p = 0.03) and tumor stage (p = 0.02) have a significant association with OS in univariate models (Table 5).
Univariate and multivariate Cox proportional hazard analyses with overall survival as endpoint.

Values in the table represent numbers of patients. Asterisks mean that the p value is statistically significant. NI: not informed; Mut: mutated; Wt: wild type; UnMeth: unmethylated; Meth: methylated; MSS: microsatellite stable; MSI-H: microsatellite instability-high; CIMP: CpG island methylator phenotype; AJCC: American Joint Committee on Cancer; TNM: tumor−node−metastasis.
Discussion
The present study was designed to characterize and describe the three main carcinogenic molecular pathways (CIN, MSI, and CIMP) involved in heterogeneity and molecular complexity of Chilean CRC patients to improve diagnose and treatment. In clinical practice, the TNM (tumor−node−metastasis) classification31,32 is used as the main prognostic tool for selecting patients for adjuvant chemotherapy. It has been described that the survival of patients in early stages exceeds 90% but in metastatic stages is near 14%. 33 In recent years, the incorporation of molecular therapies has allowed improved patients’ survival; however, due to the heterogeneity of tumors, they do not have the same effectiveness.34–37 To address this problem, in 2015 an international CRC subtyping consortium was established based on data from “The Cancer Genome Atlas” supplemented with molecular and mutational analyses. 38 Four consensus subgroups were established: CMS1 (MSI-immune), CMS2 (canonical), CMS3 (metabolic), and CMS4 (mesenchymal) with a prevalence in the international population of 14%, 37%, 13%, and 23%, respectively. This classification reflects a specific biological evolution during the development of tumors, allowing to define specific therapies for patients’ better response and survival after treatment.36,38–40 However, attending that this classification involves hundreds of genes which are difficult to implement in clinical settings, we could only subclassify the Chilean patients in four subgroups based on the three main carcinogenic pathways, with a prevalence of 15%, 38%, 30%, and 15% for MSI-H (Group 1), CIN-H (Group 2), [MSI/CIMP/CIN] low-negative (Group 3), and CIMP-H (Group 4), respectively.
CMS1 (MSI-H/CIMP-H and MSS/MSI-L/0/CIMP-H)38,40 tumors have characteristics previously reported by Jass 41 for MSI-H/CIMP-H tumors in sporadic CRC. Sessile serrated adenomas/polyps have been described as a precursor lesion for this group.40,42,43 On the other hand, the V600E activating mutation in the BRAF oncogene, constitutively activates the EGFR pathway, which as a consequence produces an inefficient response to cetuximab and/or panitumumab treatment. These patients have an intermediate prognosis 36 and a very poor survival rate after relapse. 44 In our cohort, we observed the worst overall survival for Group 1, which unlike the Guinney’s CMS1 group, we did not include patients with MSS/MSI-L/0/CIMP-H tumors. Furthermore, CMS2 tumors originate from tubular adenomas exhibiting high levels of Wnt/MYC signaling by the loss of APC tumor suppressor.39,45 Although we do not have molecular profiles that account for the activation of the Wnt/MYC pathway, we could establish that CIN-H tumors (Group 2) are associated with mutations in oncogenes such as KRAS and PIK3CA and have clinical characteristics associated with a better response to therapies related to CIN-H. On the other hand, we have reported that tumors belonging to Group 3 (low/negative) exhibit the highest rate of activating mutation in the KRAS oncogene. Therefore, these patients would probably have a low response to biological treatment. The CMS3 group, similar to our Group 3, corresponds to epithelial tumors of intermediate prognosis and whose precursor lesion has not been determined yet. 36 CMS4 tumors have been described as the subtype with the worst prognosis.36–38 They originate from serrated sessile polyps, but unlike the CMS1 subtype, they induce the activation of the transforming growth factor β (TGF-β) signaling pathway, which triggers intracellular signaling associated with the epithelial-mesenchymal transition (EMT).39,45 Unlike the CMS4 characteristics, according to Guiney’s consensus subclassification, we classified patients with CIMP-H and MSS in Group 4 regardless of CIN status, which can probably explain the observed survival differences with respect to previously described reports.23,34,36,38
In recent years, due to the increase in the aging population and advances in screening methods, it has been proposed to study CIMP-H CRC tumors with greater attention given its frequent detection in advanced ages. 46 There are contradictory results of CIMP-H tumors regarding survival and sensitivity to therapies.18,47,48 Moreover, two groups of CIMP-H tumors are distinguished according to their MSI status, possibly due to the contribution of genetic and microenvironment factors during CIMP-H tumor progression, affecting the intratumoral milieu. MSS/CIMP-H tumors are located mainly in the left colon compared with MSI-H/CIMP-H which are not, thus the interaction between CIMP tumor cells with their microenvironment would be different. Furthermore, recent studies suggest that gut microbiota composition plays a fundamental role. 49 In addition, Nosho et al. 50 have proposed that Fusobacterium nucleatum could be associated with right-sided CRC and MSI-H by an increase in reactive oxygen species and inflammatory cytokines causing MLH1 epigenetic silencing. However, F. nucleatum status was not associated with cancer-specific survival. 50 MSI-H CRC is known to be associated with the increase of tumor-infiltrating lymphocytes, elevated host systemic immune response, and a favorable prognosis.
In our cohort, we showed that patients with CIMP-H tumors had a significantly worse prognosis compared with patients with CIMP-L/0 tumors regardless of MSI status. These observations differ from other publications. They reported that MSI-H was associated with an excellent prognosis regardless of CIMP status, and the clinical behaviors of MSS tumors were affected by the presence of CIMP-H or BRAF/KRAS mutations. 51 In general, MSI-H has a good prognosis for patients only at early stages by stimulating immune response against tumor cells.52,53 The high amount of non-functional proteins as a result of promoter methylation, produces a malfunction of the cell repair proteins including MLH1, as well as silencing many tumor suppressor genes. Only one out of 25 CIMP-H patients in our cohort was MLH1-M/MSS. Also, two patients CIMP-L/0 were MLH1-M/MSS. Given the high correlation of MLH1 epigenetic silencing with MSI-H, these cases could be explained by the variability of methylation density of the analyzed promoter region (–194 to −277). The lack of complete methylation near the site of transcriptional initiation, which would allow protein expression, could explain the MSS. 54 CRC tumors with the absence of MLH1, due to promoter methylation and consequent increased MSI-H, show different behaviors at a therapeutic level, a resistance to fluoropyrimidine-type drugs (5-fluorouracil and capecitabine) is observed in stage II patients, but a greater sensitivity to irinotecan. 18
Regarding the analysis of tumor mutations, several studies have reported frequencies of mutations in KRAS, BRAF, and PIK3CA genes. KRAS ranges from 12.9% to 48.6% for Western and 20.5%−44.1% for Asian populations; BRAF 4.1%−15.9% and 1.1%–17%, and PIK3CA 3.3%–18.7% and 2.6%−13.4%, respectively. Our frequencies are 26%, 12%, and 18% for KRAS, BRAF, and PIK3CA, respectively, which is in agreement with published data.30,55–59
The association between activating mutations in the BRAF gene (V600E) and more aggressive tumors has been reported,23,60–63 showing the worst prognosis for MSS/CIMP-H/BRAFmut patients. In our cohort of MSS and MSI-H patients, four categories of tumors with CIMP and presence/absence of mutations in KRAS, BRAF, or PIK3CA genes were established, being CIMP-H/KRASmut the group with worst survival in MSS patients. However, MSI-H/CIMP-H/BRAFmut also showed worst survival rate (data not shown). This result partially agrees with Vedeld et al., 60 Kim et al., 61 and Murcia et al. 62 who reported that the group with the worst survival was MSS/CIMP-H/BRAFmut, because we only had one patient with BRAFmut in this group, we do not have enough data to confirm. On the other hand, Bae et al. 19 describe a CIMP-H group with mutated KRAS and MSS that would have a poor prognosis in agreement to our results.
Furthermore, mutated BRAF has been reported to have a poor prognosis in both early and late stages. 13 This study shows that BRAF mutations were only observed in patients with MSI-H independently of cancer progression stage. Thus, 100% (4/4) of early-stage patients and 62% (5/8) of late-stage patients were MSI-H/BRAFmut. The differences in prognosis between these groups at early or late stages were not further studied due to the small sample size. However, our preliminary studies show that 8% (4/51) of early-stage patients have mutated BRAF compared with 17% (8/48) of late-stage patients (Table 3). According to the observations that we discussed earlier, MSI-H could improve prognosis of early-stage patients; however, their prognosis would be determined by the presence of CIMP-H. It has been described that in patients with MSI-H, BRAFmut, and CIMP-H, the precursor lesion would be a sessile serrated adenoma/polyp, which would evolve to adenocarcinoma following the serrated neoplasia pathway, with high neoplastic potential. 64 A BRAF mutation would be the driver for this pathway associated with the acquisition of motility and invasiveness of the cells in the EMT process and CIMP and MSI would determine the carcinogenesis progression.
In addition, it has been reported that tumors with a high degree of tumor budding mostly present mutations in KRAS, particularly the G12D change. 65 Although in our series there are concomitant mutations between PIK3CA-exon 9 and KRAS, it was not possible to establish a relationship with survival. The molecular meaning and its therapeutic implications regarding the coexistence of KRAS and PIK3CA mutations are still under study.
Our study has been shown to be very valuable in the Chilean population for the classification of CRC, which could likely be expanded to other South American countries and perhaps other countries under similar conditions in the world. However, it has some limitations including being a single-center study, and even though our hospital has a multidisciplinary team of specialists who participate in an oncology committee (in which therapeutic behaviors are defined for patients based on the National Comprehensive Cancer Network clinical guidelines), not all patients followed their treatments at the institution. For this reason, information on adjuvant treatments could not be included. Another limitation was the low statistical power due to the small sample size in several of the subgroups, which precluded statistical analysis. Consequently, we could not show a significant difference between MSI-H/CIMP-H and MSS/CIMP-H to differentiate the prognosis between Group 1 (MSI-H) and Group 4 (CIMP-H). Furthermore, we were not able to evaluate signals associated with the activation of TGF-β that triggers the EMT process. Nonetheless, considering that there are only a few publications on molecular biomarkers associated with survival66,67 and even less in Latin-America,57–59 we believe that our findings even if limited sample size could help physicians to clarify the nature of CRC in an individual patient, determine an adequate treatment, and define prognosis.
In summary, we have distinguished four subgroups in CRC, with different intracellular signals that give each tumor a unique characterization of its biology, possible tumor evolution and prognosis, which could help for the betterment of CRC treatment. This categorization exhibits the heterogeneity and complexity of CRC, being a cornerstone of personalized medicine.
Supplemental Material
S1 – Supplemental material for Characterization of Chilean patients with sporadic colorectal cancer according to the three main carcinogenic pathways: Microsatellite instability, CpG island methylator phenotype and Chromosomal instability
Supplemental material, S1 for Characterization of Chilean patients with sporadic colorectal cancer according to the three main carcinogenic pathways: Microsatellite instability, CpG island methylator phenotype and Chromosomal instability by Ana María Wielandt, Claudia Hurtado, Mauricio Moreno C, Cynthia Villarroel, Magdalena Castro, Marlene Estay, Daniela Simian, Maripaz Martinez, Maria Teresa Vial, Udo Kronberg and Francisco López-Köstner in Tumor Biology
Supplemental Material
suplem_method_Clinicopathologic_data_26-5-2020_CH – Supplemental material for Characterization of Chilean patients with sporadic colorectal cancer according to the three main carcinogenic pathways: Microsatellite instability, CpG island methylator phenotype and Chromosomal instability
Supplemental material, suplem_method_Clinicopathologic_data_26-5-2020_CH for Characterization of Chilean patients with sporadic colorectal cancer according to the three main carcinogenic pathways: Microsatellite instability, CpG island methylator phenotype and Chromosomal instability by Ana María Wielandt, Claudia Hurtado, Mauricio Moreno C, Cynthia Villarroel, Magdalena Castro, Marlene Estay, Daniela Simian, Maripaz Martinez, Maria Teresa Vial, Udo Kronberg and Francisco López-Köstner in Tumor Biology
Supplemental Material
TableS2_26-05-20 – Supplemental material for Characterization of Chilean patients with sporadic colorectal cancer according to the three main carcinogenic pathways: Microsatellite instability, CpG island methylator phenotype and Chromosomal instability
Supplemental material, TableS2_26-05-20 for Characterization of Chilean patients with sporadic colorectal cancer according to the three main carcinogenic pathways: Microsatellite instability, CpG island methylator phenotype and Chromosomal instability by Ana María Wielandt, Claudia Hurtado, Mauricio Moreno C, Cynthia Villarroel, Magdalena Castro, Marlene Estay, Daniela Simian, Maripaz Martinez, Maria Teresa Vial, Udo Kronberg and Francisco López-Köstner in Tumor Biology
Supplemental Material
Table_S1_26-05-20 – Supplemental material for Characterization of Chilean patients with sporadic colorectal cancer according to the three main carcinogenic pathways: Microsatellite instability, CpG island methylator phenotype and Chromosomal instability
Supplemental material, Table_S1_26-05-20 for Characterization of Chilean patients with sporadic colorectal cancer according to the three main carcinogenic pathways: Microsatellite instability, CpG island methylator phenotype and Chromosomal instability by Ana María Wielandt, Claudia Hurtado, Mauricio Moreno C, Cynthia Villarroel, Magdalena Castro, Marlene Estay, Daniela Simian, Maripaz Martinez, Maria Teresa Vial, Udo Kronberg and Francisco López-Köstner in Tuular pathological classifimor Biology
Footnotes
Acknowledgements
The authors thank the collaboration of Marcela Figueroa, Ana Maria Alvarado, and Yoselin Acuña from the Department of Pathology of Clínica Las Condes for technical assistance and to Dr. Patricia Purcell from the Massachusetts Institute of Technology for critical reading and editing of the manuscript.
Author contributions
A.M.W., C.H., and M.M.C. contributed to design concept, data acquisition, data analysis, interpretation, and drafted the manuscript. A.M.W. analyzed the MSI and CIN status of CRC samples. C.H. performed somatic mutations analysis. C.V. and M.M.C. performed CIMP studies. M.C. and M.E. performed the statistical analysis. D.S. and M.M. contributed to patient recruitment. M.T.V. was responsible for the histopathological analysis of CRC samples. F.L.K. and U.K. contributed to concept design, patient recruitment, and reviewed the manuscript critically. All authors read and approved the final manuscript.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Ethics approval
This study was approved by the ethics committee from Clínica Las Condes, Santiago, Chile. All patients gave their written informed consent.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was supported by the grant (1140012) from the National Fund for Scientific and Technological Development (FONDECYT), Santiago, Chile.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.