
Abstract
Cancer stem cells can generate tumors from only a small number of cells, whereas differentiated cancer cells cannot. The prominent feature of cancer stem cells is its ability to self-renew and differentiate into multiple types of cancer cells. Cancer stem cells have several distinct tumorigenic abilities, including stem cell signal transduction, tumorigenicity, metastasis, and resistance to anticancer drugs, which are regulated by genetic or epigenetic changes. Like normal adult stem cells involved in various developmental processes and tissue homeostasis, cancer stem cells maintain their self-renewal capacity by activating multiple stem cell signaling pathways and inhibiting differentiation signaling pathways during cancer initiation and progression. Recently, many studies have focused on targeting cancer stem cells to eradicate malignancies by regulating stem cell signaling pathways, and products of some of these strategies are in preclinical and clinical trials. In this review, we describe the crucial features of cancer stem cells related to tumor relapse and drug resistance, as well as the new therapeutic strategy to target cancer stem cells named “differentiation therapy.”
Introduction
Studies have revealed that cancer stem cells (CSCs) are a rare cell population in most cancers, including blood cancers and solid tumors. CSCs, also known as tumor-initiating cells, have the abilities to self-renew and maintain their stemness traits, to differentiate into various non-stem cancer cells, and to play a crucial role in cancer propagation, metastasis, and recurrence. 1 CSCs were first identified in acute myeloid leukemia (AML) in 1997; 2 the first CSCs from solid tumors were identified in breast cancer in 2003. 3 So far, CSCs have been isolated from almost all solid tumors, including brain cancer, 4 colon cancer, 5 pancreatic cancer, 6 prostate cancer, 7 melanoma, 8 and ovarian cancer. 9 CSCs are the smallest population at the apex state of the cancer cell hierarchy, but they play the most important role in tumorigenesis.10–12 They are also highly resistant to traditional radiotherapy and chemotherapy; therefore, the tumor can recur within short periods.13,14 In addition, CSCs are considered a source of local invasion and metastasis, demonstrating the recurrence of tumors and the difficulty of achieving a complete cure.15–17 However, lower in the hierarchy, differentiated cancer progenitor cells and terminally differentiated cancer cells comprise most of the cancer cell population and do not generate tumors. 10 Therefore, this indicates the importance of CSCs and the need to target CSCs to eliminate cancer.
Cell origin and markers of CSCs
There are two hypotheses regarding the “cell origin” of CSCs. One is that CSCs are mutated adult stem cells (ASCs). This hypothesis states that the accumulation of genetic mutations in ASCs during cell division drives cancer initiation. This is most to tissues such as skin or intestine that have a high cell turnover rate. 18 The other is that CSCs are de-differentiated mutated cells. This hypothesis states that mutations in differentiated cells can cause de-differentiation, with these cells acquiring stem cell–like characteristics. Sox2 has been reported to promote de-differentiation of pancreatic cancer cells, causing them to express stem cell–like traits. 19 In the case of glioblastomas, ID4 is the key factor that de-differentiates cancer cells into glioma stem cells. 20 This hypothesis suggests that every single differentiated cancer cell has the potential to be converted to a CSC.
It becomes important to ask how could we isolate and target CSCs? CSCs can be isolated by identifying their specific cell surface biomarkers. The first identified CSCs from AML had a CD34+CD38− phenotype. 2 Since then, more specific surface biomarkers for CSCs in AML have been discovered. For instance, CD34+CD38−HLA−DR−CD71−CD90−CD117−CD123+ cells are significantly different from normal hematopoietic stem cells (HSCs). 21 In the case of solid tumors, the first CSCs isolated from breast cancer were ESA+CD44+CD24−/low cells. 3 Moreover, CD133 is a common cell surface biomarker found in brain, 4 liver, 22 lung, 23 colon, 5 and pancreatic 24 cancers. Cells in all of these solid tumors have other specific cell surface markers, such as SSEA1 for gliomas, 25 CD44 for colon cancers, 26 α2β1 for prostate cancers, 27 and ABCG2 for lung cancers. 28 Some studies have also shown that CD133 is not a specific biomarker for CSCs in gliomas. CD133− glioma cells also have CSC properties with tumor-initiating ability.29,30 However, cell surface biomarkers are still critically important for isolating CSCs, and additional therapies that target CSCs using CSC biomarkers should be developed to effectively eradicate cancer.
Signaling pathways in CSCs
To regulate adult tissue homeostasis, ASCs maintain their self-renewal properties by controlling multiple stemness signaling pathways (Wnt/β-catenin, Hedgehog/Gli, Jagged/Notch, Janus kinase/signal transducer and activator of transcription (JAK/STAT), and nuclear factor kappa beta (NF-κB)) and differentiation signaling pathways (bone morphogenetic protein (BMP) and retinoic acid (RA); Figure 1). Similar to ASCs, CSCs possess same stemness and differentiation signaling pathway networks to retain their capacity, which are frequently dysregulated.

Representative signaling pathways in cancer stem cells. Multiple stemness signaling pathways (light red area) such as Wnt, Shh, Notch, JAK/STAT, and NF-κB and differentiation signaling pathways (light blue area) such as BMP and RA regulate self-renewal and differentiation of cancer stem cells as well as in adult stem cells. Wnt signaling pathway needs the interaction of ligand and receptor to disrupt the “destruction complex” so that β-catenin translocates into nucleus to activate transcription of various stemness factors. Shh signaling pathway also needs the interaction of ligand and receptor and leads to translocation of Gli into nucleus and transactivation of stemness factors. Notch receptor generates Notch intracellular domain (NICD) through two-time proteolysis after binding with its ligand, and NICD translocates into nucleus to activate transcription of stemness factors. The Janus kinase (JAK) activated by interaction of cytokines phosphorylates signal transducer and activator of transcription (STAT), leads to translocation of STATs into nucleus and activate transcription of target stemness factors. Canonical NF-κB signaling is activated by the IKK complex. Loss of IκBα leads to the accumulation of p65-p50 dimers in the nucleus and induces transcription of target genes. As a representative differentiation signaling pathway, BMP signaling needs phosphorylation and activation of receptor by BMP ligands. The activated receptor further phosphorylates receptor-regulated Smads, and the Smads translocate into nucleus to activate transcription of differentiation factors. After interaction with retinoic acid (RA), retinoic acid receptor binds to retinoic acid response element and activates transcription of differentiation factors. Each number in this figure represents the targeted therapeutic points listed in Table 1.
A canonical Wnt/β-catenin signaling pathway requires nuclear translocation of β-catenin to sustain normal neural stem cell status. 31 When a Wnt ligand binds to and activates its Frizzled family receptor, the receptor protein disrupts the Axin-GSK3-APC “destruction complex,” leading to stabilization of the transcription factor β-catenin by preventing its degradation. Then, β-catenin translocates to the nucleus and activates transcription of downstream genes involved in stemness. However, aberrant activation of the Wnt signaling pathway, including mutations in adenomatous polyposis coli (APC), which eliminate its ability to form the “destruction complex” and constitutively activate β-catenin, is the most well-known cause of human colon carcinoma. 32 Such mutations in APCs have also been found in other malignancies such as leukemia and breast and cutaneous carcinomas.33,34 Besides APC, β-catenin mutation is the other most well-known cause that constitutively activates Wnt signaling pathway in gastric cancers and colorectal cancers.35,36 Also, R-spondin mutations and its fusion proteins and deleterious RNF43 mutations were indicated as a driver of tumor growth in the Wnt-dependent manner in colorectal cancers.37–39 In some chronic lymphocytic leukemia (CLL) cases, frequent silencing of DKK1/2, Wnt inhibiting factors, and somatic mutations in FZD5 and BCL9 were found.40,41 Mutation or deletion of Axin1, one of the intracellular components of “destruction complex” in Wnt/β-catenin signaling pathway, has been also reported to promote malignancies of several cancers, such as hepatocellular carcinomas, sporadic medulloblastomas, and colorectal cancers.42–44
The Hedgehog/Gli signaling pathway is activated by nuclear translocation of Gli in NSCs. 45 When a Hedgehog ligand binds to its cell surface transmembrane receptor, patched (PTCH) and smoothened (SMO) protein, which is suppressed by PTCH, leads to translocation of the Gli2 transcription factor to the nucleus and regulation of transcription of downstream genes relative to stemness. However, three different mechanisms of aberrant activation of hedgehog signaling pathway were found in multiple types of cancers. 46 The first one is a ligand-independent signaling pathway activation by mutations. The patients with basal cell carcinomas showed mutations in PTCH1 or SMO so that the hedgehog signaling pathway is constitutively activated at a ligand-independent manner.47–50 The second one is a ligand-dependent signaling pathway activation by autocrine manner. Overexpression or mutations in intracellular hedgehog signaling pathway components, such as SUFU, Gli1, and Gli3, are associated with tumorigenesis of medulloblastoma or pancreatic cancers.51–54 The third one is a ligand-dependent signaling pathway activation by paracrine manner. Hedgehog ligand secreted from tumor cells binds to receptor and activates hedgehog signaling pathway in stromal cells, which provides vascular endothelial growth factor (VEGF) and insulin-like growth factor (IGF) reversely to tumor cells.55,56 Furthermore, there is a reverse paracrine manner found in some B-cell lymphomas and multiple leukemia. Hedgehog ligand secreted from stromal cells activates Hedgehog signaling pathway in tumor cells, in turn leading to activation of proliferation and survival.57,58
Notch is a receptor that interacts with the Jagged ligand. After Jagged/Notch interactions, two proteolysis reactions (including γ-secretase) generate the Notch intracellular domain (NICD) that translocates to the nucleus and regulates transcription of downstream genes involved in stemness. 59 In mammals, there are four Notch receptors (Notch1, Notch2, Notch3, and Notch4) and five ligands (DLL1, DLL3, DLL4, JAG1, and JAG2). In gliomas, Jagged/Notch signaling plays an important role in crosstalk between CSCs and endothelial cells. The signaling activated by PDGF-NO-ID4 axis promotes tumor progression through increase in CSC self-renewal and tumor vasculature. 60 Mutations in Notch receptor activate the Notch downstream signaling pathway and transactivate gene expressions involved in stem cell signaling in a Jagged ligand-independent manner. Notch1 mutations activate Jagged/Notch signaling pathway and promote tumorigenesis in breast cancer or T-lineage acute lymphoblastic leukemia (T-ALL).59,61,62 Besides, loss-of-function mutations in FBXW7, which decrease turnover of the NICD, were found in some cases of T-ALL patients.63,64 Gain-of-function mutations in Notch2 were reported in diffused large B-cell lymphomas.65,66 Moreover, accelerated Notch4 receptor activity and loss or downregulation of Notch signaling regulator Numb have been reported in CSCs in breast cancer.67,68
JAK/STAT signaling also plays an important role in both adult tissue homeostasis and tumorigenesis. JAK is activated by interactions between ligands such as cytokines and growth factors and their cell surface receptors. Activated JAK phosphorylates STAT, and phosphorylated STATs translocate to the nucleus as heterodimers or homodimers where the downstream genes are transactivated. 69 However, 9% of patients with hepatocellular carcinoma showed missense mutations in JAK1, resulting in activation of JAK/STAT signaling pathway and cytokine- and receptor-independent growth. 70 In addition, JAK2-V617F mutations have been found responsible for hematopoietic malignancies, and this point mutation overactivates the JAK/STAT signaling pathway to promote cytokine-independent growth.71–74 Furthermore, there are other types of mutations in JAK2, such as K539L,75,76 T875N, 77 and deletion of IREED. 78 In acute megakaryoblastic myeloid leukemia, JAK3 mutations on A572V, V722I, or P132T have been identified. 79 Although, rare amplifications in STAT5A/B locus have been identified in prostate cancer. These amplifications increase translocation of STAT5 into nucleus and messenger RNA (mRNA) expression, leading to promotion of tumor growth. 80
NF-κB family contains five members, such as p65 (RelA), NF-κB1 (p105/p50), NF-κB2 (p100/p52), RelB, and c-Rel. The precursors p105 and p100 cleave to active subunits p50 and p52. 81 NF-κB canonical signaling pathway is generally activated by various stimuli, including lipopolysaccharide (LPS), tumor necrosis factor-alpha (TNF-α), or interleukin-1 (IL-1). Without stimulation, p65-p50 dimers bind to the inhibitor of κB proteins (IκBs), retaining the localization of NF-κB at the cytoplasmic at its steady state. While the receptors are activated by stimuli, IκBα is phosphorylated and induces its ubiquitination proteasomal degradation by the IκB kinase (IKK) complex, which comprised one non-catalytic regulatory subunit IKKγ (a.k.a NF-κB essential modifier (NEMO)) and two kinase subunits IKKα and IKKβ. After loss of IκBα, NF-κB is accumulated in nucleus, further binds to DNA and promotes transcription of target genes which involve in inflammation, anti-apoptosis, survival, and proliferation.82–86 Furthermore, aberrant and constitutive activation of NF-κB signaling pathway also regulates cancer promotion, metastasis, and angiogenesis.87–90 Next-generation sequencing analysis revealed that amplifications of c-Rel, IKKβ, and NEMO and mutations on upstream factors Carma1, Bcl10, and Malt1 genes have been found in some lymphomas and breast cancers.91,92 Besides, overexpression of growth factor receptors, such as epidermal growth factor receptor (EGFR) and Her2, also could activate NF-κB signaling pathway in cancers.93,94 In case of glioblastoma, NF-κB-dependent transition of proneural to mesenchymal subtype promotes radiotherapy resistance and poor prognosis. 95 NF-κB also maintains tumor growth–promoting macrophage phenotype (M2) instead of tumor growth–repressing macrophage phenotype (M1).96,97
To date, many types of inhibitors of stemness signaling pathways have been developed, such as β-catenin inhibitors and ligand antagonists targeting Wnt/β-catenin signaling, SMO and Gli inhibitors targeting Hedgehog signaling, γ-secretase inhibitors and ligand antagonists targeting Notch signaling, JAK and STAT inhibitors targeting JAK/STAT signaling, and NF-κB signaling inhibitors (Table 1). Most of these inhibitors and antagonists are in the preclinical and clinical trial phase I/II/III, and one of them has already been approved by the FDA.
Inhibitors or antagonists used for targeting stemness signaling pathways.
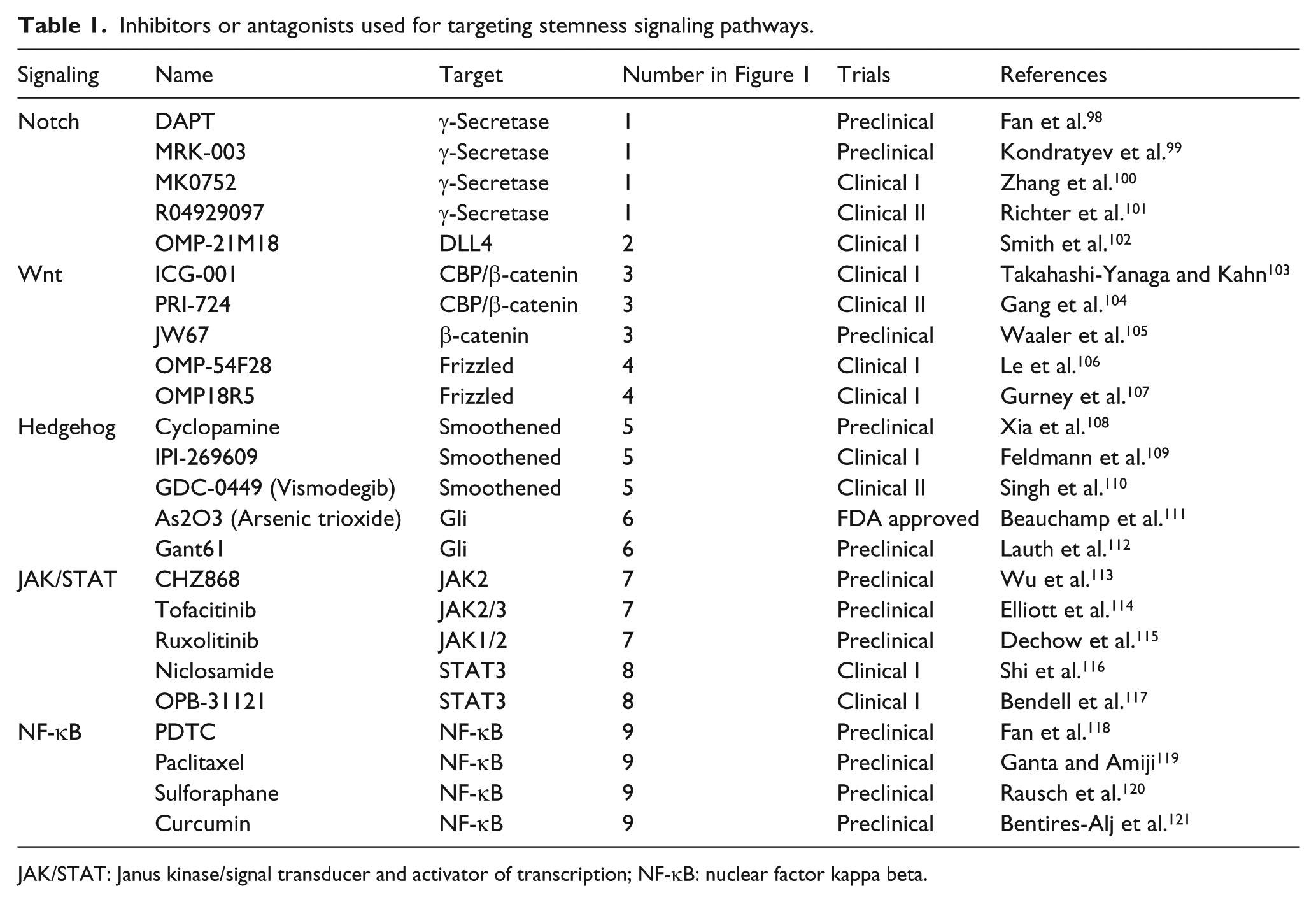
JAK/STAT: Janus kinase/signal transducer and activator of transcription; NF-κB: nuclear factor kappa beta.
Induction of CSC differentiation
ASCs and CSCs have the potential to differentiate into multiple types of cells, including lineage-differentiated cells and non-tumorigenic-differentiated cancer cells upon exposure to specific differentiation signals (Figure 1).
RA, a small lipophilic derivative of vitamin A, is a well-known inducer of differentiation and proliferation in normal stem cells. By interacting with RA, retinoic acid receptor (RAR) binds to the retinoic acid response element (RAREs) on DNA as a heterodimer with retinoid X receptor (RXR), leading to transcription of genes that regulate differentiation. 122 During early vertebrate organogenesis, RA functions as an essential factor for intercellular signaling. 123 A pharmaceutical form of RA named tretinoin was developed and is already used as an inducer in differentiation therapy for patients with acute promyelocytic leukemia (APL). 124 It is also known as all-trans retinoic acid (ATRA), a carboxylic acid form of vitamin A. Induction of differentiation by treatment with ATRA can increase sensitivity to therapies and reduce CSC motility and tumorigenicity by blocking angiogenic cytokines in glioblastomas. 125 ATRA treatment to breast cancer stem-like cells, MCF7/C6, leads to cell differentiation, reduced invasion and migration, and increased sensitivity to anticancer treatment. 126 However, in some glioma study, ATRA induces differentiation of CSC, but their differentiation was not induced perfectly, thereby glioma CSC appears again. 127
BMP signaling is the most powerful inducer of differentiation. BMP is a secreted glycoprotein belonging to the transforming growth factor beta (TGF-β) superfamily. It is a ligand that binds to receptor BMPR2, which phosphorylates the neighbor receptor BMPR1. Then, phosphorylated BMPR1 induces phosphorylation of receptor-regulated Smads (R-Smads: including Smad1, Smad5, and Smad8). Furthermore, the phosphorylated R-Smads form heterodimers with co-Smad (Smad4) and translocate to the nucleus, where downstream genes are transcriptionally regulated. 128 Most genes transcriptionally activated or suppressed by BMP signaling are regulators of terminal differentiation. 129 BMP signaling has two side functions in cellular differentiation in adult tissues: BMP signaling functions as both a suppressor of Wnt/β-catenin signaling pathway and a cell fate determinant by inducing differentiation. These phenomena often occur in the skin, central nervous system, and intestine.130–132 At an early phase in embryonic development, BMP signaling promotes the growth of neural stem cells. In a later phase, BMP signaling blocks proliferation of neural precursors and induces their differentiation. 133 In the case of cancers, impaired BMP signaling or overexpression of BMP antagonists promotes tumorigenesis in primary tumor sites or cancer cell colonization in metastatic sites. For example, the tumor stromal cells expressing high level of Gremlin 1 (an antagonist of BMP) create a CSC niche that allows glioma CSCs to maintain their self-renewal ability by inhibiting BMP signaling. 134 BMP7 secreted from endogenous neural precursor cells inhibits proliferation, self-renewal, and tumorigenicity of glioma CSCs. 135 A recent study also demonstrated that a variant type of BMP7 decreases tumor growth, angiogenesis, and invasion by inducing differentiation of glioma CSCs. 136 Thus, induction of differentiation by activating BMP-associated signaling is a promising strategy for cancer treatment. A previous study showed that chromatin modifications of the BMPR2 promoter inhibit the expression of BMPR2 and that restoration of chromatin in the BMPR2 promoter or ectopic transduction of BMPR1B induces differentiation of CSCs and decreases tumorigenicity in glioblastomas. 137
Inhibition of CSC differentiation
During early embryonic development, differentiation signaling is less effective than stemness signaling. Hence, decreased differentiation signaling is as important as maintaining self-renewal capacity of ASCs and CSCs.
How do ASCs and CSCs inhibit differentiation? Inhibition of differentiation/DNA-binding (ID) family genes is the most well-described function of the proteins that inhibit lineage differentiation. 138 There are four members of the ID family, all of which are basic helix-loop-helix (bHLH) transcription factors without a DNA-binding domain. ASC and CSC traits are often maintained by ID proteins. When ID proteins are downregulated, a homodimer or heterodimer of the E protein binds to E-boxes and regulates transcription of genes involved in lineage differentiation. 138 Highly expressed ID proteins in normal stem/progenitor cells are known to negatively regulate transcription of genes involved in lineage differentiation and cell-cycle inhibition so that cells can maintain their stemness properties. In contrast, low levels of ID protein expression induced cell differentiation. When ID proteins were absent, mice showed lethality, with defects in immune and vascular cells and a reduction in self-renewal and proliferation of neural stem/progenitor cells. Similarly, decreased ID proteins were associated with the loss of CSC properties, along with a reduction in angiogenesis and tumorigenesis. 138 In case of malignant high-grade gliomas, ID proteins are involved in various phenotype and capacities of glioma CSCs. For example, ID1 activates Wnt/β-catenin and Hedgehog/Gli stemness signaling pathways, leading to maintenance of self-renewal and tumorigenesis of glioma CSCs. 139 EGFR amplification or oncogenic EGFR variant III mutant gene activates glioma CSC generation and angiogenesis through ID1 and ID3 upregulation. 140 ID4 is a forceful reprogramming factor that convert glioma cells or immortalized astrocytes to glioma CSCs by dedifferentiation. 20
MicroRNAs (miRNAs) are known to negatively regulate gene expression by pairing perfect or imperfect complementary sequences within the 3′-untranslated region (UTR) of mRNA. They are noncoding RNA molecules ~22 nucleotides in length that are abnormally expressed in various cancers. There are two kinds of miRNAs in cancers: oncogenic miRNAs (oncomiRs) and tumor-suppressive miRNAs. OncomiRs are generally elevated in cancers and are reported to increase cancer cell progression, survival, and metastasis. MiR-148a is a well-known oncomiR that inhibits tumor suppressor genes, MIG6 and BIM, and indirectly increases EGFR expression and phosphorylation in CSCs of glioblastomas. 141 In addition, a recent study showed that delivery with synthesized spherical nucleic acids from miR-182 (182-SNAs) resulted in reduction of tumor burden and increase in mice survival by inducting differentiation of glioma CSCs. 142
Strategies to target CSCs
In the last few decades, numerous cancer therapies have been developed. However, malignancies are still a threat because of resistance to drug therapy and irradiation. The major cause of this resistance is CSCs.
Several therapeutic strategies to eliminate CSCs have been identified. Cell surface biomarkers such as CD44, CD47, and CD133 are considered the primary targets for CSC treatment. CD44 is specifically expressed in leukemia stem cells; it is not present in normal HSCs. 143 Treatment with antibodies against CD44 significantly decreased CSCs in AML-bearing mice. CD47 is also highly expressed in acute lymphoblastic leukemia (ALL) and thus could be a specific target for treatment of CSCs in ALL. 144 As described above, CD133 is a cell surface biomarker for many kinds of cancers, including glioblastomas and colon and prostate cancers. Patients with high levels of CD133 expression have a poor prognosis. 145 Thus, several studies have been conducted to target CSCs expressing high levels of CD133.146,147
In addition, stemness signaling pathways, stemness biomarkers, epigenetic alterations, and even the microenvironment could be potential therapeutic or predictive targets in CSCs. Stemness signaling pathways such as Wnt/β-catenin, Hedgehog, Jagged/Notch, JAK/STAT, and NF-κB have been shown to be abnormally activated in various malignancies. Thus, inhibiting or reversing these abnormal stemness signaling pathways might be a promising strategy for targeting CSCs (Table 1). Blockade of Notch signaling was shown to significantly reduce the CD44+CD24−/low CSC subpopulation and brain metastasis in breast cancers. 148 Pharmaceutic or antibody treatment that targets Wnt/β-catenin signaling shows strong anti-tumor effects.149,150 Decoying Myc, the transcriptional target of Wnt/β-catenin signaling, reduced the growth of CSCs and induced their differentiation. 151 In addition, modification of the aberrant Wnt signaling pathway has been proposed as a potential method for targeting CSCs. 152 Targeting of Hedgehog signaling in CSCs has also been accomplished with an Hedgehog antagonist and SMO inhibitor.153,154 Impaired NF-κB signaling leads to polarization of M2 to M1 phenotype, which in turn increases inflammatory cells and reduces tumor progression.95,96 Nestin and CD146 have been shown to play important roles in tumor metastasis, suggesting the possibility of predicting early recurrence. 155 In addition, some natural products can affect the epigenetic alterations in CSCs and inhibit cancer initiation and progression. 156 Besides, a new therapeutic strategy was recently proposed to target the microenvironment of CSCs for cancer treatment. A novel antagonist of CXCR4, a receptor for CXCL12 that increases in tumors, induced CSC differentiation and reduced tumor cell growth in a preclinical glioblastoma multiforme (GBM) model. 157
Conclusion
Recently, targeting CSCs is emerging as an important therapeutic strategy.158–160 Because targeting cell surface biomarkers of CSCs or key stemness signaling pathways might impair normal stem cells, inducing CSC differentiation offers promise as a relatively safe cancer treatment. BMP-mediated differentiation of CSCs is a promising strategy. Although the BMP signaling cascade is abnormal in glioblastomas because of promoter hypermethylation in CSCs, demethylation of the promoter or forced expression of BMPR2 can induce CSC differentiation and reduce tumorigenicity. 136 Leukemia was the first tumor subjected to anticancer research for developing a differentiation therapy. 161 As mentioned earlier, ATRA has been used for the treatment of APL, in which differentiation is inhibited by fusion genes with the RA receptors (promyelocytic leukemia-RA receptor α (PML-RARα)).162,163 Therefore, whether other cancers show similar effects is worth examining. Although alkylating antineoplastic agents (e.g. cyclophosphamide and busulfan) are the most popular anticancer drugs, these agents only kill fast-growing cancer cells, but not relatively quiescent CSCs. Differentiation therapy might induce CSCs into terminally differentiated cancer cells, and then, these cells could be treated with alkylating agents or a combination of the two therapies. In recent years, many studies have shown that cells that undergo epithelial-to-mesenchymal transition (EMT) acquire stemness features from several tumor types.164–168 Therefore, differentiation therapies that induce mesenchymal-to-epithelial transition (MET) have been proposed as a new alternative. 169 Although induction of MET has a high risk of metastatic colonization at distant sites, this method can still be considered an attractive therapeutic strategy, as it reduces aggressive tumorigenic CSCs. To eradicate cancer and prevent recurrence, differentiation strategies require additional research and development.
Footnotes
Acknowledgements
We are grateful to all members of the Cell Growth Regulation Laboratory for their helpful discussions.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by grants from the National Research Foundation (NRF), funded by the Ministry of Science, ICT and Future Planning (2015R1A5A1009024); from the Next-Generation Biogreen21 Program (PJ01107701); and from Korea University. X.J. (Xun) was supported by grants from the General Program of the National Natural Science Foundation of China (No. 81572891).