
Abstract
Gastric cancer is the third leading cause of cancer-related death worldwide, but the mechanisms of gastric carcinogenesis are not completely understood. Recently, the role of cholinergic neuronal pathways in promoting this process has been demonstrated. Our aim was to extend these studies and to evaluate, using an in vitro model of tumorspheres, the effect of acetylcholine on human gastric cancer cells, and the role of acetylcholine receptors and of the nitric oxide pathway, in this effect. The gastric cancer cell line MKN-45 of the diffuse type of gastric cancer was cultured in the presence of acetylcholine, or different agonists or inhibitors of muscarinic and nicotinic acetylcholine receptors, or nitric oxide donor or inhibitor of the nitric oxide pathway, and the number and size of tumorspheres were assessed. The expression of cancer stem cell markers (CD44 and aldehyde dehydrogenase) was also evaluated by immunofluorescence and quantitative reverse transcription polymerase chain reaction. We showed that acetylcholine increased both the number and size of tumorspheres and that this effect was reproduced with both muscarinic and nicotinic acetylcholine receptors agonists and was inhibited by both receptor antagonists. The nitric oxide donor stimulated the tumorsphere formation, while the nitric oxide synthesis inhibitor inhibited the stimulatory effect of acetylcholine. Moreover, acetylcholine increased the expression of stem cell markers on gastric cancer cells. These results indicate that acetylcholine induces the stem cell properties of gastric cancer cells and both muscarinic and nicotinic receptors and a nitrergic pathway might be involved in this effect.
Keywords
Introduction
Gastric cancer (GC) is the third leading cause of mortality from cancer in men in the world, 1 and despite the progress in treatment, its prognosis remains poor, especially for the diffuse type of this cancer, with an overall 5-year survival rate not exceeding 25%. 2 The mechanisms of gastric carcinogenesis are not completely understood, although the role of infection by Helicobacter pylori, as well as of some dietary factors, has been recognized. 3 Recently, the implication of the parasympathetic nervous system (PNS) and cholinergic pathways in the pathogenesis of GC has been demonstrated. Using three different mice models of gastric carcinogenesis, Zhao et al. have shown that stomach denervation by surgical vagotomy or local injection of neurotoxic agent (botulinum toxin) inhibits the development of gastric tumors and attenuates the progression of pre-neoplastic lesions into cancer. They have also shown, using an in vitro model of gastric organoids, that acetylcholine (ACh) muscarinic 3 (M3) receptor can modulate gastric stem cells. 4 In a more recent study, 5 the same group studied the ACh signaling in transgenic mouse models and showed that nerve- and DCLK1+ tuft cell–derived ACh upregulated nerve growth factor (NGF) which, in turn, stimulated gastric innervation, tumor initiation, and TRK/YAP-mediated gastric stem cell expansion, and that NGF blockage or tuft cell ablation inhibited tumorigenesis. The role of ACh in gastric carcinogenesis was also explored by Yang et al., 6 who showed that ACh, through its M3 receptor, promotes the invasion and migration of GC cells through the M3R/AMPK/MACC1 oncogenic pathway.
All these data strongly indicate that ACh promotes gastric carcinogenesis, but the exact mechanisms (i.e. receptors and signaling pathways involved) are not completely understood. In particular, the possible implication of nicotinic ACh receptors in this process is not clear, although the promoting role of tobacco smoking in GC has been suggested by some epidemiological studies and meta-analyses,7,8 and the capacity of alpha-7 nicotinic receptor to stimulate the migration of GC cells has been shown in vitro. 9 Moreover, the effect of ACh on gastric cancer stem cells (CSCs), as previously suggested, 4 needs to be further elucidated. Indeed, CSCs represent a unique subpopulation of cells within the tumors, which possess the ability to initiate tumor growth and sustain tumor self-renewal, and which are responsible for cancer recurrence. 10 The existence of CSCs in GC was demonstrated in human GC on the basis of tumorigenic functional assays showing their capacity to initiate tumorsphere formation in vitro, and these cells were essentially characterized by the expression of the cell surface marker CD44 and by high aldehyde dehydrogenase (ALDH) activity.11–14
The nitric oxide (NO) pathway is involved in several effects of ACh. It has been shown that ACh may induce the proliferation of cancer cells by stimulating nitric oxide synthase (NOS) activity 15 and that NOS may activate the muscarinic receptors in different cancer cell lines. 16 It has been also shown that endogenous NO regulates the stemness properties in colon CSCs through cross regulation of several cellular signaling pathways. 17 It is now recognized that NO generated at the inflammatory site may contribute to the initiation and progression of different cancers. 18 All these data suggest that the effects of ACh on GC cell could be, at least in part, mediated by NO.
Therefore, our aim was to study, using an in vitro model of tumorspheres, the effect of ACh on human gastric CSCs, the role of both muscarinic and nicotinic ACh receptors in this effect and the mechanistic role of the NO pathway.
GC cell line culture
Adenocarcinoma gastric epithelial cells MKN-45 (from RIKEN, BRC Cell Engineering Division, Japan) of the diffuse type of GC were cultured in Dulbecco’s Modified Eagle Medium (DMEM)/F12 media, with 10% fetal bovine serum and 50 µg/mL vancomycin (all from Invitrogen, Cergy-Pontoise, France) at 37°C in a 5% CO2 humidified atmosphere, as previously described. 14
Tumorsphere formation
Cells were recovered and 5000 cells were plated on non-adherent 12-well culture plates (coated with a 10% polyHEMA (Sigma, Saint-Quentin-Fallavier, France) solution in absolute ethanol and dried overnight at 56°C). After plating, the cells were incubated for 5 days at 37°C in a serum-free medium consisting of DMEM–F12 GlutaMAX supplemented with 20 ng/mL of epidermal growth factor, 10 ng/mL of basic fibroblast growth factor, 1:100 N2 supplement 100×, 0.3% glucose, 5 µg/mL of gentamicin, and 50 IU/mL of penicillin (all from Invitrogen). 14 The tumorspheres were identified using an IN Cell Analyzer 2200 microscope system with 20× objective, and the number and size of tumorspheres in each well were quantified using the System snapshot file in IN Cell Analyzer 2200/6000 software. A tumorsphere was defined as a non-adherent cluster of more than 10 cells, as previously described. 13 The size of tumorspheres was evaluated by measuring the major axis of each tumorsphere using ImageJ software.
Neurotransmitters and cell treatment
ACh, bethanechol (selective ACh muscarinic receptor agonist), dimethylphenylpiperazinium (DMPP; selective ACh nicotinic receptor agonist), atropine (selective ACh muscarinic receptor antagonist), hexamethonium (selective ACh nicotinic receptor antagonist), sodium nitroprusside (SNP; NO donor), and NG-nitro-L-arginine methyl ester (L-NAME; NO synthesis inhibitor), all purchased from Sigma, were used at the concentrations ranging from 0.1 to 10 µM. In different sets of experiments, the cells were cultured in adherent condition in the presence of different neurotransmitters for 5 days, and then the cells were recovered, washed, and cultured without any agent for 7–10 days in non-adherent condition to favor the formation of tumorspheres.
Analysis of the expression of stem cell and epithelial–mesenchymal transition markers
The expression of stem cell (CD44, ALDH1A1, SOX2, KLF4) and epithelial–mesenchymal transition (EMT; Vimentin, Snail1, Zeb1) markers was studied on adherent cells, incubated in the presence of 1 µM ACh for 5 days.
The expression of CD44, ALDH1A1, Vimentin, Snail1, Zeb1, SOX2, and KLF4 was analyzed by real-time quantitative polymerase chain reaction (RT-qPCR) as previously described.14,19 In brief, total cellular RNAs were extracted using the Trizol reagent (Invitrogen) according to the manufacturer’s protocol. Total RNA was converted to cDNA using the QuantiTect Reverse Transcription (RT) kit (Qiagen, Courtaboeuf, France) according to the manufacturer’s recommendations. Quantitative polymerase chain reaction (PCR) was performed using the SYBR Green qPCR Master Mix (Promega, Charbonnières-les-Bains, France) and specific primers (Cat. No. QT00998333 for CD44, QT0001328 for ALDH1A1, QT00095795 for Vimentin, QT00010010 for Snail1, QT00020972 for Zeb1, QT0023760 for SOX2, and QT00061033 for KLF4, purchased from Qiagen), to analyze the expression of CD44 and ALDH1A1 genes. Real-time PCRs were run on an Applied Biosystems ABI StepOnePlus Real-Time PCR System. Each sample was measured in duplicate. Gene expressions were normalized relative to HPRT1 genes using the comparative Ct method.
In addition, the CD44 expression and ALDH activity were analyzed by immunofluorescence. To this aim, the live adherent cells were incubated in phosphate-buffered saline (PBS) containing anti-CD44 antibody (clone G44-26; BD Biosciences, Le Pont-de-Claix, France) at 4°C for 30 min, washed twice with PBS buffer, and then analyzed using an IN Cell Analyzer 2200 microscope system with 20× objective. The activity of ALDH was measured by enzymatic assay using the ALDEFLUOR Kit (STEMCELL Technologies, Grenoble, France), as previously described. 13
Analysis of the role of the NO pathway in the effect of ACh on GC cells
Cells cultured in adherent condition were treated for 5 days with ACh (1 µM), SNP (NO donor, 0–1000 µM), or ACh+ L-NAME (NO synthase inhibitor, 10 µM). After 5 days of treatment, the cells were recovered and cultured in non-adherent condition to evaluate their capacity to form the tumorspheres.
Statistical analysis
Quantitative values represent the mean of three or more independent experiments, each performed in at least three or more biological replicates, and are expressed as mean ± standard deviation (SD). Statistics were performed using Mann–Whitney test, Kruskal–Wallis test, or two-way non-parametric analysis of variance (ANOVA) test, depending on the type of comparison, using SPSS 16.0F software (SPSS Inc., Chicago, IL, USA).
Results
ACh induces the CSC properties of GC cells of diffuse type
The primary objective of this study was to determine whether ACh is capable of inducing CSC phenotype in GC cells as assessed by their capacity to form tumorspheres. To this aim, MKN45 cells were treated for 5 days in adherent conditions in the presence of ACh at different concentrations, before being cultured in non-adherent condition for 7–10 days. The impact of ACh was evaluated by the ability of the pretreated cells to form tumorspheres (their number and size).
ACh at the concentrations of 0.1 and 1 µM significantly increased the number of tumorspheres (p < 0.0001 as compared to the control cells, 10 < n < 14), but this effect was not observed at the highest ACh concentration of 10 µM (Figure 1(a)–(c)). ACh also significantly increased the size of tumorspheres and this effect was observed at all concentrations (p < 0.05, as compared to the control cells, 16 < n < 32; Figure 1(a), (b), and (d)).

Effect of ACh on tumorsphere formation by GC cells in vitro. GC cells of diffuse type (MKN45) were cultured in adherent condition for 5 days in the presence of ACh at the concentrations of 0.1, 1, or 10 µM and then cultured without ACh for 7–10 days in non-adherent condition to form tumorspheres. Microscopic images (magnification 200×) of tumorspheres in control conditions (a) and in the presence of 1 µM ACh (b). The number (c) and size (d) of tumorspheres (mean ± SE) in control conditions and after treatment with ACh (10 < n < 14). ACh at the concentrations of 0.1 and 1 µM increased the number of tumorspheres (***p < 0.001 as compared to the control cells, Kruskal–Wallis test), but this effect disappeared at the highest ACh concentration of 10 µM. ACh at all concentrations increased the size of tumorspheres (d) (*p < 0.05 as compared to the control cells, Kruskal–Wallis test, 16 < n < 32). Scale bars: 100 µM.
ACh induces the CSC properties of GC cells via muscarinic and nicotinic receptors
The secondary objective was to study, using the pharmacological approach, the involvement of muscarinic and/or nicotinic receptors in the ACh stimulatory effect on the CSC properties of GC cells. First, we showed that the selective ACh muscarinic receptor agonist, bethanechol, increased the number and size of tumorspheres at a concentration of 1 µM (*p < 0.05, **p < 0.003, ***p < 0.001 as compared to the control cells; n = 7), but not at the highest (10 µM) concentration used (Figure 2(a) and (b)). Conversely, the stimulatory effect of ACh on the number and size of tumorspheres was significantly reduced by the muscarinic receptor inhibitor, atropine (*p < 0.05, **p < 0.01, ***p < 0.001; n = 7; Figure 2(c) and (d)). Second, we showed that DMPP, a selective nicotinic receptor agonist, significantly increased the number and size of tumorspheres at all concentrations (0.1, 1, and 10 µM) (*p < 0.05, **p < 0.003, ***p < 0.0003; n = 6; Figure 2(e) and (f)). Conversely, hexamethonium, a nicotinic receptor antagonist, inhibited at a concentration of 100 µM the stimulatory effect of ACh on tumorsphere formation by diminishing the number and size of tumorspheres as compared to ACh-stimulated cells (*p < 0.05, **p < 0.03, ***p < 0.0001; 6 < n < 8; Figure 2(g) and (h)).

Effect of nicotinic and muscarinic ACh receptors on tumorsphere formation by GC cells in vitro. MKN45 GC cells were cultured in adherent condition for 5 days in the presence of bethanechol (selective ACh muscarinic receptor agonist) (a, b), atropine (selective ACh muscarinic receptor antagonist) (c, d), dimethylphenylpiperazinium (DMPP; selective ACh nicotinic receptor agonist) (e, f), or hexamethonium (selective ACh nicotinic receptor antagonist) (g, h) at the concentrations of 0.1, 1, or 10 µM and then cultured without any agent or with ACh at 1 µM for 7–10 days in non-adherent condition to form tumorspheres. The number (a, c, e, g) and size (b, d, f, h) of tumorspheres (mean ± SE) in control conditions and after treatment with different agents (6 < n < 8) (*p < 0.05, **p < 0.01, ***p < 0.001 as compared to the non-treated cells, Kruskal–Wallis test). Both agonists (bethanechol and DMPP) at the concentrations of 0.1 and 1 µM increased both the number (a) and size (b) of tumorspheres and this effect was abolished at the highest concentration of 10 µM. DMPP also increased the size of tumorspheres at all concentrations (d). Both ACh receptor antagonists (atropine and hexamethonium) at the concentration of 100 µM inhibited the stimulatory effect of ACh (1 µM) on tumorsphere formation by diminishing the number and size of tumorspheres as compared to ACh-stimulated cells (**p < 0.01, ***p < 0.0001, two-way non-parametric ANOVA test) (e, f, g, h). These results indicate that both nicotinic and muscarinic receptors are involved in the stimulatory effect of ACh on tumorsphere formation by GC cells.
The stimulatory effect of ACh on CSC phenotype of GC cells is partly mediated by the NO pathway
We next aimed to identify the possible mechanisms involved in the ACh effects on gastric CSC phenotype and focused on the possible implication of the nitrergic pathway.
First, we showed that the NO donor, SNP, increased the number and size of tumorspheres at the concentrations of 1, 10, and 100 µM. However, at the highest concentration of 1 mM, SNP completely inhibited the capacity of GC cells to form tumorspheres (*p < 0.05, n = 4; Figure 3(a)). Conversely, the pretreatment of cells with an NO synthesis inhibitor (L-NAME) significantly inhibited the number of tumorspheres formed by ACh-stimulated cells as compared to L-NAME non-treated cells (*p < 0.05 as compared to ACh-treated cells, ***p < 0.001, n = 7; Figure 3(b)). Altogether, these results indicate that ACh may act, at least in part, via stimulation of the nitrergic pathway.
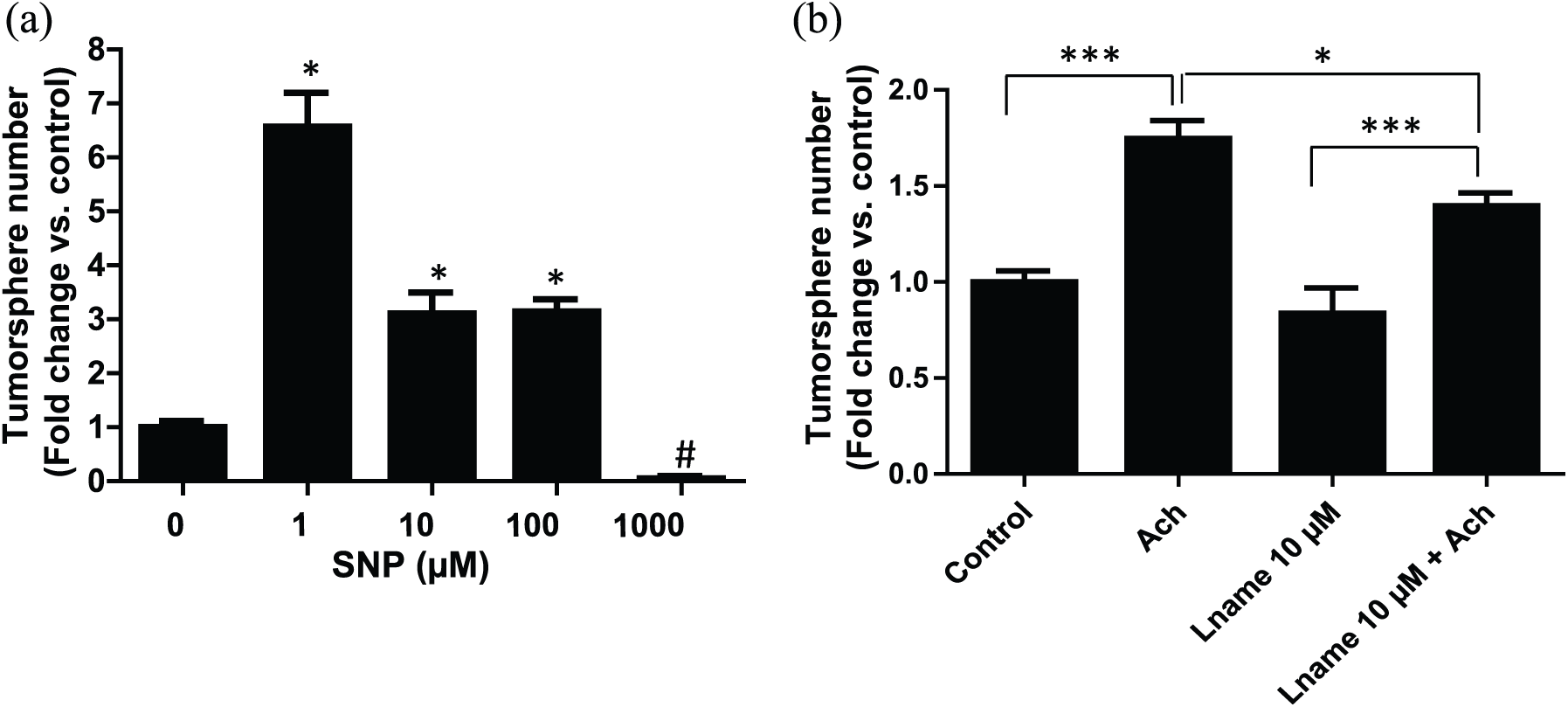
Effect of NO on tumorsphere formation by the GC cells. Cells were incubated for 5 days in the media with or without sodium nitroprusside (SNP), known as an NO donor. Effect of SNP on tumorsphere formation of GC cells depended on concentration. At low concentrations up to 100 µM, SNP significantly increased the number of tumorspheres as compared to the control condition (*p < 0.05, n = 4, Mann–Whitney test). In contrast, at the highest concentration of 1000 µM, SNP completely inhibited the capacity of GC cells to form tumorspheres (#p < 0.01 as compared to the control conditions, Mann–Whitney test) (a). In the second part of the experiment (b), the cells were pretreated with ACh (1 µM) and/or an NOS inhibitor L-NAME (10 µM) for 5 days and then cultured in non-adherent condition to form tumorspheres. Inhibition of NOS using L-NAME led to the diminution of the number of tumorspheres formed by ACh-stimulated cells as compared to L-NAME non-treated cells (*p < 0.05, ***p < 0.001 as compared to ACh-treated cells, n = 7, two-way non-parametric ANOVA test). These results indicate that the nitrergic pathway may be involved in the stimulatory effect of ACh on tumorsphere formation by GC cells.
ACh induced CSC and EMT expression markers in GC cells
The self-renewal of stem cells and the EMT are the key events often activated during cancer invasion and metastasis, and associated with the acquisition of stem cell properties.20,21 Therefore, we desired to see whether the effect of ACh was associated with the modulation of the expression of CSC and EMT markers in our model. Two stem cell markers were analyzed: CD44, identified as a marker of stem cells of several types, highly expressed in many cancers, including GC, and ALDH1A1, one of the main isoforms of the ALDH isoenzyme family responsible for the activity, recognized as the most specific marker of gastric CSC.11,13,22 The expression of the three major EMT markers, Vimentin, Snail1, and Zeb1, and two self-renewal markers of stem cells, SOX2 and KLF4, was analyzed. 19 Using RT-qPCR, we showed that ACh significantly increased the CD44 and ALDH1A1 mRNA expression levels in GC cells after 24 h and especially after 48 h (*p < 0.05, **p < 0.001, ***p < 0.0001, n = 3; Figure 4(a) and (b), respectively). We confirmed this finding on protein level by showing an increased CD44 expression and ALDH activity of GC cells after their incubation with ACh (Figure 5). We also showed that ACh increased the mRNA expression levels of other stem cell markers (SOX2 and KLF4) and also EMT markers (Snail1, Zeb1, and Vimentin) (*p < 0.02, **p < 0.0001, n = 3; Table 1). These results indicate that ACh induces a CSC phenotype by stimulating the expression of markers of CSC, stem cell self-renewal, and EMT.

Effect of ACh on CSC markers’ expression by GC cells. Cells were cultured in adherent condition without or with ACh (1 µM) for 5 days. The expression of CD44 and ALDH was analyzed by RT-qPCR and is expressed as relative mRNA expression fold change as compared to the controls. An increased expression of CD44 (a) and ALDH (b) in ACh-treated cells, as compared to control cells, was observed after 24 h and especially after 48 h of treatment (*p < 0.05, **p < 0.001, ***p < 0.0001 by Mann–Whitney test, n = 3).

Effect of ACh on CD44 and ALDH expression by immunofluorescence. Cells were cultured in adherent condition without or with ACh (1 µM) for 5 days. The CD44 protein expression and ALDH activity were analyzed by immunofluorescence. An increased expression of CD44 (a) and ALDH (b) in ACh-treated cells, as compared to the control cells, was observed.
Effect of acetylcholine (ACh) on mRNA expression of stem cell markers (SOX2, KLF4) and epithelial–mesenchymal transition markers (Zeb1, Vimentin, Snail1) by gastric cancer cells, after 8, 24, and 48 h of exposure to ACh (1 µM), determined by RT-qPCR, expressed as median (min–max) of mRNA expression fold change as compared to controls (n = 3).

Significant difference as compared to controls by Mann–Whitney test, *p < 0.02 and **p < 0.0001.
Discussion
ACh is the major neurotransmitter of the digestive tract and regulates various physiological processes in the stomach, such as acid and hormonal secretion. Besides its physiological role, ACh and cholinergic pathways could contribute to the development of gastric diseases, and in particular of GC.4,5 Indeed, it has been shown that ACh can directly stimulate GC cell proliferation, 23 invasion, and migration 6 in vitro. Furthermore, Zhao et al. 4 showed that stomach vagal denervation inhibited GC development and the genetic knockout of M3 receptor–suppressed tumorigenesis in mice. Our results add to these findings by showing that, besides the muscarinic pathways, the nicotinic pathways might also be involved and, additionally, that the ACh effect on CSC phenotype may be, at least in part, mediated by the NO-dependent pathway.
One of the original findings of our study is that ACh can induce a CSC phenotype in GC cells of the diffuse type. Indeed, most of the previous studies focused on the effect of ACh on the intestinal type of GC.4,5 Our study shows that, similarly to what has been observed in the intestinal type of GC, ACh also favors the CSC phenotype of GC cells of the diffuse type as attested by its capacity to increase the number and size of tumorspheres. This effect was in part exerted via the activation of muscarinic receptors since bethanechol reproduced the ACh effect and atropine blocked it. However, we also showed the contribution of the nicotinic-dependent pathways in this effect. This is a particularly important finding since, while the implication of muscarinic receptors in gastric carcinogenesis has been highlighted by several studies,4–6,23,24 there are much less data on the potential role of nicotinic receptors in this process. Indeed, the association between GC and tobacco smoking has been demonstrated by epidemiological studies and meta-analyses showing an increased risk of GC in smokers as compared to non-smokers with a relative risk of about 1.6 and a dose–response relationship.8,25 Different pathogenic mechanisms have been proposed, the major one being a direct pro-cancerous effect of several carcinogens included in the tobacco. Nicotine by itself does not appear to have a direct carcinogenic effect, but some data suggest that the stimulation of nicotinic receptors (nicotinic acetylcholine receptor (nAChR)), which are known to participate in cellular adhesion and migration, may promote cell growth and angiogenesis and inhibit the drug-induced apoptosis of cancer cells.26,27 We did not specifically look for the nicotinic receptor subtype involved, and future studies will be necessary to identify it. The potential candidates may include an alpha-7 nAChR, already shown to be involved in the migration of GC cells 9 and in their sensitivity to different chemotherapeutical agents.28,29
The factors involved in the induction of CSC phenotype by ACh in diffuse-type GC cells may include the modulation of CSC markers, such as CD44 and ALDH, and also the enhancement of EMT gene expression by ACh, as demonstrated in our study. CD44 is recognized as a phenotypic marker of CSC, but it also has known functional significance since CD44 siRNA has been shown to drastically inhibit cell survival and tumorsphere formation. 12 EMT enables the cancer cells to disseminate and recent evidence suggested that the induction of EMT is associated with the acquisition of stem cell properties.14,20
Another important finding of our study is the identification of the NO pathway as contributing to the induction of the CSC phenotype by ACh. Indeed, we showed that NO at low concentrations favored the ACh effect, while L-NAME prevented this effect. Similar pro-proliferative properties of NO have been already reported, as well as the deleterious effects of high doses of NO. 30 It is worth to notice that low doses of NO are produced during chronic inflammation which could amplify the ACh effects and favor gastric carcinogenesis. In GC, the chronic inflammation driven by H. pylori leads to the expression of host inflammatory genes which in turn activate several enzymes, including NOS2, the high expression of which has become an important determinant of gastric carcinogenesis associated with H. pylori infection. 18
Our results can also be considered in line with those recently obtained in the experimental models of intestinal type of GC where ACh was shown to mediate its effects via NGF-dependent pathway. 5 It remains to be shown whether NO could also contribute to the modulation of NGF secretion by ACh.
In conclusion, our results further confirm the role of ACh in gastric carcinogenesis and indicate its capacity of inducing stem cell phenotype in GC cells of diffuse type of GC. They also point to the possible role of the nitrergic pathway and of nicotinic receptors in this effect, which could become new therapeutic targets in GC in the future.
Footnotes
Acknowledgements
Michel Neunlist and Tamara Matysiak-Budnik share co-senior authorship.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was supported by the French National Society of Gastroenterology (SNFGE), la Ligue Contre le Cancer, SANTEDIGE Foundation, and the Ministry of Science and Technology in Vietnam (grant no. 55/B2017-TNA-55).