
Abstract
Multiple factors including tumor heterogeneity and intrinsic or acquired resistance have been associated with drug resistance in lung cancer. Increased stemness and the plasticity of cancer cells have been identified as important mechanisms of resistance; therefore, treatments targeting cancer cells independent of stemness phenotype would be much more effective in treating lung cancer. In this article, we have characterized the anticancer effects of the antibiotic Nigericin in cells displaying varying degrees of stemness and resistance to anticancer drugs, arising from (1) routine culture conditions, (2) prolonged periods of serum starvation. These cells are highly resistant to conventional anticancer drugs such as Paclitaxel, Hydroxyurea, Colchicine, Obatoclax, Wortmannin, and LY294002, and the multidrug-resistant phenotype of cells growing under prolonged periods of serum starvation is likely the result of extensive rewiring of signaling pathways, and (3) lung tumorspheres that are enriched for cancer stem-like cells. We found that Nigericin potently inhibited the viability of cells growing under routine culture conditions, prolonged periods of serum starvation, and lung tumorspheres. In addition, we found that Nigericin downregulated the expression of key proteins in the Wnt canonical signaling pathway such as LRP6, Wnt5a/b, and β-catenin, but promotes β-catenin translocation into the nucleus. The antitumor effects of Nigericin were potentiated by the Wnt activator HLY78 and by therapeutic levels of the US Food and Drug Administration–approved drug Digitoxin and its novel synthetic analog MonoD. We believe that Nigericin may be used in a co-therapy model in combination with other novel chemotherapeutic agents in order to achieve potent inhibition of cancers that display varying degrees of stemness, potentially leading to sustained anticancer effects.
Introduction
Lung tumors display high intratumoral heterogeneity that is associated with chemoresistance.1,2 As a result, the tumor invariably relapses in patients with lung cancer after few cycles of chemotherapy. Multiple factors including architectural heterogeneity caused by differentiation, 3 epidermal growth factor receptor (EGFR) mutation heterogeneity, 4 and metabolic heterogeneity 5 may affect response to anticancer agents. Lung tumors are composed of cancer cells that display varying degrees of proliferation and stemness and are found in specific microenvironmental niches caused by factors such as hypoxia, paracrine signaling, and non-cancer-associated cells.6–9 These subpopulations of cancer cells are often referred as cancer stem cells (CSCs) or cancer stem-like cells (CSLCs) and are consistently more resistant to conventional chemotherapy. 10 While CSCs/CSLCs were initially considered a distinct subpopulation of cells that if eliminated could cure cancer, it has now been demonstrated that CSCs/CSLCs and non-CSCs can interconvert into each other. 11 As a consequence, anticancer agents targeting only the CSCs/CSLCs subpopulations may not be successful. In order to improve the response to chemotherapy, it is essential to identify new drugs or drug combinations that can target a broad spectrum of cancer cells with varying degrees of proliferation and stemness found within microenvironmental niches in tumors, and not just specific subpopulations of cancer cells.
We used distinct in vitro culture conditions for drug screening in human H460 lung that led to three different cell populations that displayed varying degrees of proliferation, stemness, and chemosensitivity in a manner that mimics intratumoral heterogeneity found in vivo: (1) routine culture conditions (RCCs), (2) prolonged periods of serum starvation (PPSS), and (3) floating tumorspheres (FTs). FTs compared to cells growing under RCCs are also resistant to PX, HU, and CX and express different stemness-associated markers. 12 Compared to cells growing under RCCs, cells growing in PPSS proliferate slower and are strongly resistant to conventional anticancer drugs such as Paclitaxel (PX), Hydroxyurea (HU), Colchicine (CX), Obatoclax (OBT), Wortmannin (WT), and LY294002 (LY). 13 The multidrug-resistant phenotype of cells growing under PPSS is likely the result of extensive rewiring of signaling pathways involved in the regulation of multidrug resistance and survival proteins such as MDR1, ABCG2, and Bcl-2. 14 Therefore, distinct in vitro culture conditions may in part mimic the intratumoral heterogeneity found in vivo in tumors with the additional advantage that specific cancer cells subpopulations (phenotypes) can be identified as target of the drug tested.
In this study, we evaluated in our in vitro model of tumor heterogeneity the antitumor effects of Nigericin (NIG), an antibiotic derived from Streptomyces hygroscopicus that works by acting as an H+, K+, and Pb2+ ionophore. While NIG has been shown to specifically target CSCs in nasopharyngeal carcinoma 15 and to suppress metastasis in colorectal cancer, 16 its effect in lung cancer cells is largely unknown. NIG was found to downregulate the Wnt signaling pathway in non-cancer cells such as human foreskin fibroblast 17 and human embryonic kidney cells, 18 but not in cancers. Wnt signaling is recognized as an important player in the development and metastasis of lung cancer and has emerged as a promising target to develop agents toward cancer prevention and therapy. 19 We have recently shown that Digitoxin (DIG), a cardiac glycoside that inhibits the Na+/K+-ATPase, and its synthetic analog MonoD (MD) were able to decrease the viability of H460 cells growing under RCCs, as well as under PPSS. 13 More importantly, both NIG 20 and DIG or MD, 13 at low (nM) concentrations, have effects on key signaling pathways (see section “Discussion”) that may be exploited to enhance drug sensitivity when used in combination with other drugs. The aim of this study was to test the anticancer activity of NIG alone or in combination with DIG or MD in human H460 lung cancer cells displaying varying degrees of proliferation and stemness.
Materials and methods
Drugs
DIG and HLY78 were obtained from Sigma–Aldrich (St. Louis, MO), and NIG and Wnt-1 were purchased from VWR (Radnor, PA). DIG and MD (β-D-digitoxose) were stored as stock solutions (10 mM) in dimethyl sulfoxide (DMSO) in glass containers at −20°C. NIG and HLY78 were stored as stock solution (25 and 10 mM, respectively) in DMSO at −20°C. Wnt-1 was reconstituted in sterile H2O containing bovine serum albumin (0.1%) as protein carrier, aliquoted and stored at −80°C. MD was synthesized using a methodology previously described. 21 Final dilutions were freshly prepared in culture media before use. The control experiment contained only (~0.01%) of DMSO at the highest concentration.
Cell culture
The human lung epithelial NCI-H460 cancer cell line was obtained from American Type Culture Collection (Manassas, VA). This cell line is considered highly resistant to chemotherapy.
22
NCI-H460 cells were cultured in RPMI 1640 supplemented with 5% fetal bovine serum (FBS), 2 mM
Subcellular fractionation
For determination of β-catenin translocation into the nucleus, cell fractionation was performed by the REAP (Rapid, Efficient And Practical) method 24 with slight modifications. Briefly, H460 cells growing in 100-mm Petri dishes were washed twice with ice-cold phosphate-buffered saline (PBS), scraped from culture dishes on ice using a plastic cell scraper, and collected in 1.5-mL micro-centrifuge tubes in 1 mL of ice-cold PBS. After centrifugation of a “pop-spin” for 10 s in an Eppendorf table-top microfuge, the supernatants were removed and the cell pellets were resuspended in 700 µL of ice-cold 0.1% NP40 substitute in PBS and triturated 5 times using a p1000 micropipette. 300 µL of the lysate was removed as “whole cell lysate” and 60 µL of 6× Laemmli sample buffer sodium dodecyl sulfate (SDS) sample buffer, reducing, (Boston Bioproducts Cat# BP-111R) was added to it, and then kept on ice until the sonication step. The remaining material was centrifuged for 10 s, 400 µL of the supernatant was removed as the “cytosolic fraction,” and 80 µL of 6× Laemmli sample buffer was added to this fraction. The remaining pellet was resuspended in 1 mL of ice-cold 0.1% NP40 substitute in PBS and centrifuged as above for 10 s and the supernatant was discarded. The pellet (~20 µL) was resuspended with 300 µL of 1 × Laemmli sample buffer and designated as “nuclear fraction.” Nuclear fractions and whole cell lysates that contained DNA were sonicated using microprobes with a Model 505 Sonic Dismembrator (Fisher Scientific, Hampton, NH) at 20% level, twice for 5 s each. All fractions were heated at 95°C for 5 min. For western blotting, samples of 30, 30, and 30 µL of whole cell lysate, cytoplasmic, and nuclear fractions, respectively, were loaded into polyacrylamide gel electrophoresis–sodium dodecyl sulfate (PAGE-SDS).
Short-term antiproliferative effects (MTT assay)
For RCCs, cells (~2000 cells/well) were plated in 96-well cell-culture microplates (Costar, USA) and incubated overnight in complete media (CM)—RPMI 1640 supplemented with 5% FBS, 2 mM
Colony-forming assay
Colony-forming assay was performed according to Rafehi et al. 25 Briefly, 200 cells/well were plated in 6-well plates and allowed to adhere overnight. Cells were then treated with drugs at the indicated concentration or with vehicle alone for 72 h in CM (Dulbecco’s Modified Eagle’s Medium (DMEM) containing 5% FBS). After drug exposure, cells were incubated with CM for 7–10 days (media was changed every 72 h). Cells were fixed with 3.7% formaldehyde for 15 min, stained with 0.01% crystal violet, and photographed. Colonies were counted using ImageJ Software (version 1.48, http://imagej.nih.gov/ij/).
Flow cytometry
For flow cytometry analysis, cells grown in 100-cm Petri dishes were treated with drugs or vehicle for 24 h and collected by tripsinization, washed twice with PBS, and fixed overnight in 70% ethanol at 4°C. Cells were then washed twice with PBS, treated with DNAse-free RNAse (100 µg/mL), and stained with propidium iodide (50 µg/mL). The cells were then analyzed using the ACEA NovoCyte 2060 (ACEA Biosciences, San Diego, CA) and NovoExpress (version 1.0.2) cell cycle analysis software. The cell cycle distribution is shown as the percentage of cells containing G0/G1, S, and G2/M DNA as identified by propidium iodide staining.
Migration and invasion assays
Migration and invasion assays were performed using cells serum-starved overnight. For migration assays, H-460 cells (1.05 × 105 cells) were seeded in 8-µm pore size Transwell® Inserts (Corning®, NY) in RPMI 1640 (without FBS) with or without drugs (DMSO as control) or NIG (1 µM). Inserts were placed in 24-well plates with RPMI 1640 containing 10% FBS in the presence of DMSO (control) or NIG (1 µM). After 24 h, cell migration was quantified. Nonmigratory cells were removed by gently removing the media and washing with PBS. Following manufacturer’s instructions, the membranes containing migratory cells were stained with Hema 3 Stat Pack (Fisher, Kalamazoo, MI). Migratory cells were photographed (five pictures for each treatment) under light microscope at 20× objective and counted. Invasion assays were performed in the similar way except that cells were seeded in matrigel-coated 8-µm pore size Transwell Inserts (Corning, NY) and invasion was quantified after 48 h. All experiments were independently repeated three times.
Tumorsphere assay
Cells growing in poly(2-hydroxyethyl methacrylate) (polyHEMA)-coated plates as floating lung tumorspheres (LTs) were obtained as previously described. 12 For viability assays, LTs were collected in 15-mL Falcon tubes, centrifuged at 700 r/min × 3 min and resuspended in fresh SFM. In order to plate the same number of cells, this cell suspension was split in 1-mL aliquots. Vehicle or drugs were added to each aliquot, and then 150-µL cell suspension was loaded into each microwell (in a 96-well plate) and incubated for 72 h. Cell viability was evaluated by the Cell Counting Kit-8 (CCK-8) assay (Dojindo Molecular Technologies, Inc., Rockville, MD).
Western blotting
Preparation of cell lysates and western blotting were performed as described previously. 26 Antibodies for LRP6, phospho-LRP6, Wnt5a/b, Naked2, Axin, β-catenin, Lamin A/C, α-tubulin, and β-actin were purchased from Cell Signaling Technology, Inc. (Danvers, MA). Peroxidase-conjugated secondary antibody was purchased from Santa Cruz Biotechnology (Dallas, TX). The immune complexes were detected by chemiluminescence and quantified using analyst/PC densitometry software (Bio-Rad Laboratories, Hercules, CA).
Statistical analysis
The IC50 (drug concentrations inhibiting cell growth by 50%) were determined by interpolation from the dose-response curves using a sigmoidal logistic three-parameter equation. Each point represents the mean ± standard deviation (SD) of quadruplicate or sextuplicate wells (see figures for details). Curve fitting and all pairwise multiple comparison procedures (analysis of variance (ANOVA) and Student–Newman–Keuls method) and Student’s t-test have been done using SigmaPlot (version 11.0) software.
Results
NIG inhibits proliferation and clonogenicity of H460 lung cancer cells
The antiproliferative effect of NIG was evaluated in RCCs. Figure 1 shows that NIG decreased the viability of H460 cells in a concentration-dependent manner. The anticlonogenic activity of NIG was evaluated by the colony-forming assay. NIG at 0.1 µM decreased the formation of colonies by approximately 70% and 1 µM was able to completely prevent colony formation (Figure 1(b)).

Nigericin decreases the viability of H460 cells. (a) Concentration-dependent effect of Nigericin. H460 cells were incubated with the increasing concentrations (0–50 µM) of Nigericin for 72 h. Cell viability was determined by the MTT assay. Data (mean ± SD) are representative of two independent experiments performed by sextuplicates. (b) Colony-forming assay. Cells were incubated with the indicated concentration of Nigericin for 72 h followed by incubation in drug-free media for ~9 days. Data (mean ± SD) are representative of two independent experiments performed by triplicates.
NIG inhibits migration and invasion of H460 lung cancer cells
The inhibitory effects of NIG on the migratory and invasive properties of H460 lung cancer cells were evaluated by trans-cell migration and invasion assays in the presence of NIG (1 µM). NIG significantly inhibited both migration and invasion (Figure 2).

Nigericin (NIG) inhibits migration and invasion of H460 lung cancer cells. H460 cells were seeded at 1.05 × 105 cells/well in the presence of vehicle (control, DMSO 0.0005%), or NIG 1 µM allowed to migrate (a) or invade (b) toward FBS for 24 or 48 h, respectively. Migratory or invasive cells on the bottom of the membrane were quantified as described in the assay protocol. Data (mean ± SD) are representative of three independent experiments.
Wnt-1 and HLY78 do not antagonize NIG effects on cell viability
To test whether NIG directly targets the Wnt signaling pathway, we performed co-incubation studies using the Wnt agonist Wnt-1. H460 cells were incubated wit NIG alone (1 µM) or in the presence of Wnt-1 (10, 50, or 100 ng/mL) for 72 h. Figure 3(a) shows that co-incubation with Wnt-1 does not prevent NIG-induced decrease in cell viability in cells growing under RCCs. However, HLY78, a small molecule activator of the Wnt/β-catenin signaling pathway inhibited cell viability and potentiated the effect of NIG (Figure 3(b)).
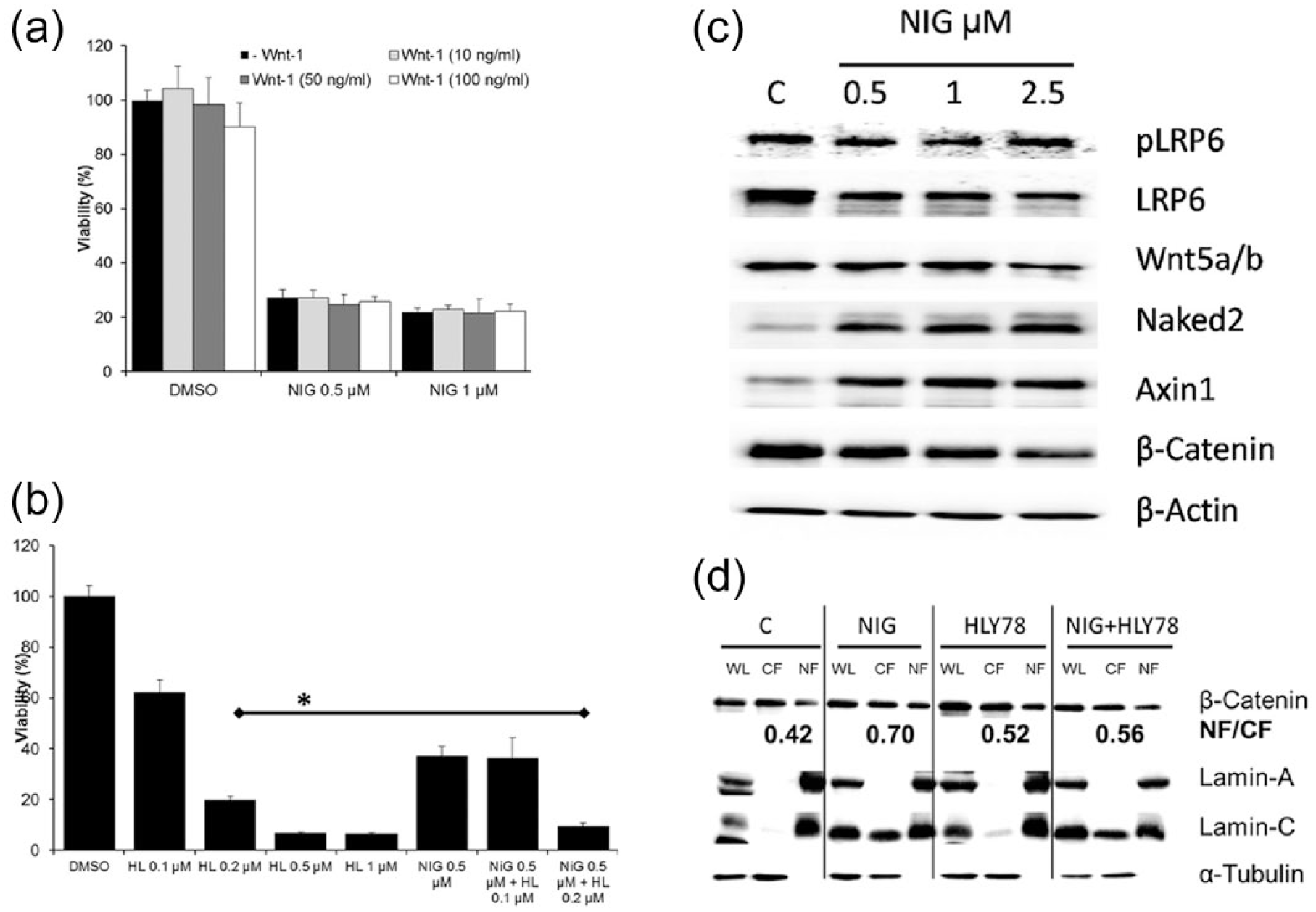
Nigericin potentiates the effect of the Wnt activator HLY78 and downregulates the Wnt signaling pathway, but promotes β-catenin translocation into the nucleus. (a) Wnt agonist Wnt-1 does not affect cell viability and Nigericin effects on cell viability. H460 cells were incubated with NIG alone (0.5 or 1 µM) or in the presence of increasing concentration of Wnt-1 (10, 50, or 100 ng/mL) for 72 h. Control cells were incubated with DMSO alone. Cell viability was determined by the MTT assay. (b) Wnt activator HLY78 decreases cell viability in a concentration-dependent manner and potentiates the effect of NIG. H460 cells were incubated with increasing concentrations (0.1–1 µM) of HLY78 for 72 h (first 5 columns). In parallel, cells were incubated with NIG alone (0.5 µM) or NIG (0.5 µM) plus 0.1 or 0.2 µM HLY78 (last 3 columns) for 72 h. Control cells (c) were treated with equivalent concentration of DMSO. (c) Nigericin downregulates key proteins of the Wnt signaling pathway. H460 cells were incubated with increasing concentrations (0.5–2.5 µM) of Nigericin for 24 h. Control cells (c) were treated with equivalent concentration of DMSO. Equal amount of proteins (30 µg) were loaded into each well, separated by PAGE-SDS and detected with specific antibodies by immunoblotting. (d) NIG promotes translocation of β-catenin into the nucleus. H460 cells were incubated with NIG alone (1 µM) or in the presence of HLY78 (0.1 µM) for 24 h. Whole cell lysates (WL), cytoplasmic fraction (CF), and nuclear fraction (NF) were separated by the REAP method, and 30 µL of each fraction were loaded into each well, separated by PAGE-SDS and detected with specific antibodies by immunoblotting. The number in bold indicates the NF/CF ratio of β-catenin measured by densitometry (ImageJ). The data for NIG are representative of four independent experiments, and the data for HLY78 or NIG+HLY78 are representative of two independent experiments.
NIG downregulates the Wnt signaling pathway but promotes β-catenin translocation into the nucleus
The expression of several key proteins of the Wnt signaling pathway was evaluated by Western Blot in H460 cells treated with DMSO (control) or NIG (0.25, 1, and 2.5 µM) for 24 h. As shown in Figure 3(c), NIG induced a clear concentration-dependent decrease in the expression of LRP6, Wnt5a/b, and β-catenin, while levels of Axin and Naked2 increased even with low levels of NIG (0.5 µM). This result demonstrates that NIG downregulates the expression of important protein of the canonical Wnt signaling pathway. In order to test whether NIG inactivates Wnt signaling, we evaluated the effect of NIG on β-catenin translocation into the nucleus. H460 cells were treated with DMSO (control) or NIG (1 µM) for 24 h. We used HLY78 as positive control. After treatment, whole cell lysates, cytoplasmic fractions, and nuclear fractions were separated by the REAP method (see section “Materials and methods”), and the expression of β-catenin was evaluated by Western Blots. Figure 3(d) shows that while NIG decreased the global expression of β-catenin (consistent with the data presented in Figure 3(c)), the relative amount of this protein was elevated in the nuclear fraction. Due to fact that in the REAP method protein concentrations are not measured prior to electrophoresis, we quantitated the relative increase in β-catenin in the nuclei by determining the nuclear fraction/cytoplasmic fraction (NF/CF) ratio by densitometry using ImageJ. It is expected that if β-catenin translocates to the nuclei, the β-catenin NF/CF ratio will increase. Indeed, HLY78 (0.1 µM), that was used as a positive control, promoted β-catenin translocation into the nucleus and increased this ratio from 0.42 to 0.52. Our data showed that NIG increased this ratio to 0.70. The purity of the nuclear fraction was evaluated by assessing the expression of Lamin A/C. During apoptosis, Lamin A/C (70 kDa) is specifically cleaved to a large (40–45 kDa) and a small (28 kDa) fragment. In DMSO-treated cells, full-length Lamin A/C (70 kDa) was almost exclusively detected in the nuclear fraction, but in NIG-treated cells, the global levels of full-length Lamin A/C, as well as its 28-kDA cleaved fragment, were elevated. The latter was detected in the cytoplasmic fraction in NIG-treated cells. The purity of the cytoplasmic fraction was evaluated by assessing the expression of α-tubulin that, as expected, was almost exclusively detected in the whole cell and cytoplasmic fraction, but not in the nuclei.
NIG decreases the viability of lung cancer cells growing as tumorspheres
We next evaluated the antiproliferative effect of NIG in LTs. It is important to mention that for viability assays, the tumorspheres were plated in uncoated 96-well microplates and therefore untreated cells rapidly reattach (see arrows in Figure 4(a)). In contrast, loss of cellular integrity, as well as failure to reattach, is evident after 24 h treatments with NIG 5 µM. LTs were incubated with NIG (0, 1, 5, or 10 µM) for 72 h and viability was evaluated using the CCK-8 assay. NIG decreased the viability of LTs in a concentration-dependent manner (Figure 4(b)).

Nigericin decreases the viability of lung tumorspheres. (a) Representative images of LTs showing loss of cellular integrity in LTs treated with NIG for 24 h. Magnification = 20× and bars = 200 µM. The thick arrows show cells that rapidly reattach to the uncoated plates. (b) The bars indicates the mean OD of sphere-forming H460 cells after treatments with different concentrations of Nigericin measured by the CCK assay.
NIG has antiproliferative effect in cells growing under different culture conditions, which is potentiated by therapeutic concentrations of DIG and MD
The effect of NIG was tested in cells growing under RCCs, PPSS, and in LTs treated for 72 h. NIG alone was found to be more potent in cells growing under RCCs (IC50 < 1 µM; Figures 1(a) and 5(a) as compared to cells growing under PPSS (IC50 ~ 5 µM; Figure 5(b)). In LTs, NIG at 0.5 µM was able to decrease the viability by ~50% (Figure 5(b)). Both DIG and MD at 20 nM each were able to potentiate the effect of NIG on cell viability (Figure 5(a)–(c)). The effect was more pronounced in cells growing under PPSS and in LTs and showed that a regimen with low concentration of NIG (~1 µM) in combination with therapeutic concentration of DIG or MD can have a similar antiproliferative effect on H460 cells, independent of culture conditions.

Digitoxin and MonoD potentiates the antiproliferative activity of Nigericin in cells growing. (a) under routine culture conditions. (b) under prolonged serum starvation conditions. (c) Digitoxin and MonoD potentiates the antiproliferative activity of Nigericin in the clonogenic assay. Data (mean ± SD) are representative of two independent experiments performed by triplicates. (d) in lung tumorspheres. Cells were incubated for 72 h with the indicated concentrations of Nigericin alone or Nigericin + DIG or MonoD. Cell viability was determined by the MTT assay for adherent cells (a, b) and by the CCK assay for floating tumorspheres.
NIG as single agent inhibited the ability of H460 cells to form colonies (Figure 5(d)), and this effect was potentiated when NIG was used in combination with DIG (20 nM) or MD (20 nM) (Figure 5(b)). Flow cytometry evaluation did not detect any effect on cell cycle distribution in H460 cells treated for 24 h with low concentration of NIG alone (0.1 µM) or in combination with therapeutic levels of DIG (20 nM) or MD (20 nM) (Figure S1).
Discussion
In this study, we used NIG both as a lead compound and in combination with DIG or MD to evaluate the chemosensitivity of human H460 lung cancer cells growing under different culture conditions. NIG is a K+/H+ ionophore derived from S. hygroscopicus that has been recently shown to overcome multidrug resistance of cancer cells 27 and to target CSCs.15,28–30 However, a recent study found that stem-like ovarian cancer cells expressing high levels of ABC drug transporters were largely resistant to ionophore antibiotics (IAs) such as salinomycin and NIG suggesting that IAs alone cannot eliminate certain CSLCs.31,32 NIG also inhibits cellular processes associated with increased stemness such as metastasis and epithelial mesenchymal transition (EMT) in colorectal 16 and breast cancer cells. 29 In agreement with those previous findings, we found that NIG decreased the viability and clonogenicity of H460 cells (Figure 1), as well as their ability to migrate and invade (Figure 2). NIG inhibited invasion more than migration. This is likely due to the fact that in these assays, migratory and invasive cells are typically evaluated after drug exposure for 24 and 48 h, respectively. Therefore, prolonged exposure to drugs (48 h) may slightly overestimate the effect of NIG on invasion. Regardless of this issue, NIG significantly inhibited migration within 24 h. The activation of the Wnt signaling pathway has been associated with enhanced migratory and invasiveness properties in non-small-cell lung cancer. 33 In non-cancer cells, NIG was shown to inhibit the Wnt signaling pathway17,18 that is important for stem-cell maintenance,34,35 but the role of NIG-mediated Wnt inhibition in cancer cells is not known. To further elucidate the role of Wnt signaling in NIG effects on cell viability, we performed a series of experiments using Wnt-1 and HLY78. Addition of Wnt-1 had little effect on viability of H460 cells and did not prevent the effects of NIG (Figure 3(a)). Furthermore, western blots demonstrated that Wnt-1 had no effect on the expression of β-catenin (data not shown). The lack of effect of Wnt-1 could be due to the presence of Wnt-1 in the FBS and/or as result of autocrine secretion that may have fully activated the Wnt pathway. Although the levels of Wnt-1 in FBS are not known, in healthy human serum of Wnt-1 were 233 ng/mL (IQR 62-1756). 36 For this reason, we used HLY78, a small molecule activator of the Wnt/β-catenin signaling pathway. HLY78 promotes LRP6 phosphorylation and Wnt signaling transduction by targeting the DIX domain of Axin and potentiating the Axin–LRP6 association. 37 HLY78 was found to decrease the viability of H460 cells in a concentration-dependent manner and surprisingly, potentiated rather than antagonized the effect of NIG (Figure 3(b)). At the molecular level, NIG decreased the expression of LRP6, phospho-LRP6, and β-catenin and increased the expression of Axin1 and Naked2 in a concentration-dependent manner, indicating that NIG targets the Wnt canonical signaling pathway in lung cancer cells (Figure 3(c)). Axin1 is a negative regulator 38 and Naked2 is an inducible antagonist of Wnt signaling. 39 Although NIG decreased the global levels of β-catenin, it did not prevent its translocation into the nucleus. In fact, in NIG-treated cells, β-catenin was found to be elevated in the nuclear fraction compared to untreated cells (Figure 3(d)), suggesting that NIG may actually promote nuclear translocation and activate rather than inactivate the Wnt signaling pathway. It is important to mention that in previous studies, the downregulatory effect of NIG on Wnt signaling pathways has been evaluated by western blots from whole cell lysates.17,18 Our findings explain why HLY78 potentiate rather than antagonize the NIG effects on cell viability. At the molecular level, HLY78 promoted β-catenin translocation into the nucleus but contrary to NIG did not significantly decrease total levels of this protein. We suggest that NIG promotes translocation of β-catenin into the nucleus by a different mechanism. Similar results were observed in Calu-3 cells: both NIG and HLY78, when used as single drug, decreased the viability but showed a potentiated effect when used in combination (Figure S2), further supporting a model in which NIG, despite downregulating the total levels of β-catenin, actually increases the relative nuclear amount in the nuclear fraction and activates the Wnt pathway. Our hypothesis is in agreement with recent findings that have demonstrated that both activation and inactivation of Wnt can inhibit cellular proliferation. 40
We also demonstrated that therapeutic levels of DIG (20 nM) was able to potentiate the effect of relative low concentration of NIG (0.5–1 µM). For instance, in cells growing under RCCs and as LTs, a combination of NIG (0.5 µM) and DIG or MD (20 nM) was able to inhibit cell viability by almost 90% (Figure 5(a) and (c)), and in cells growing under PPSS, a combination of NIG (1 µM) and DIG or MD (20 nM) was able to inhibit cell viability by more than 75%. Given that increased cancer stemness is associated with resistance, such potent synergy may be important from the perspective of developing novel co-therapy regimens that are able to significantly inhibit tumors independent of stemness status.
The mechanism by which NIG, DIG, and MD as single agents kill cancer cells is poorly understood. NIG at relatively high concentration (2.5–7.5 µM) may trigger apoptosis41,42 or necrosis 43 by depletion of intracellular potassium due to its ability to increase K+ efflux. In this study, we found that in addition to its effects on Wnt signaling pathway, NIG upregulated the expression and cleavage of Lamin A/C (Figure 3(d)) that is consistent with activation of apoptosis. Direct effect on mitochondrial function was reported at 1 µM in Jurkat cells. 30 It is possible that NIG at low concentrations (125–500 nM) may induce apoptosis by a different mechanism such as modulation of the Wnt signaling pathway as has been reported in chronic lymphocytic leukemia cells. 18 NIG has the potential to be used in combination with other anticancer agents: it enhanced the cytotoxicity of mafosfamide 20 and sensitized glioma cells to TRAIL (Tumor necrosis factor–Related Apoptosis-Inducing Ligand)-induced apoptosis. 27 However, DIG is a well-characterized Na+/K+-ATPase inhibitor. 44 At relatively high concentrations (0.5–5 µM), DIG has the ability to inhibit the Na+/K+-ATPase pump, but at low concentrations (10–100 nM) may activate several transcriptional regulatory cascades via the Na+/K+-ATPase signalosome. 45 We recently found that DIG downregulates the expression of proteins involved in chemoresistance, survival, and stemness maintenance such as MDR1, ABCG2, Bcl-2, pAKT, and key proteins of the Wnt signaling pathway. 14 We have summarized the potential interplay of the effects exerted by NIG and DIG in Figure 6.

Effects of Digitoxin and Nigericin at micromolar and nanomolar concentrations. Digitoxin at micromolar concentration inhibits the Na+/K+-ATPase, and increases (Na+) and decreases (K+) intracellular concentration. At nanomolar concentration, Digitoxin activates the Na+/K+-ATPase signalosome activating downstream signaling pathways. 45 Nigericin at micromolar concentration decreases intracellular (K+) concentration by its ability to increase K+ efflux 41 and at nanomolar concentrations inhibits the expression of key proteins of the Wnt signaling pathway 18 and promotes translocation of β-catenin into the nucleus.
Understanding the mechanism by which the combination of NIG and DIG or MD exerts anticancer activity with similar potency in cells with different degrees of stemness will be an important step in identifying new druggable targets. However, using drug combinations that target distinct pathways in an effort to observe additive antitumor effects is not a trivial approach—it is more likely that multiple mechanisms and microenvironmental factors dictate the mechanism by which each single drug decreases the viability of cancer cells. For instance, we recently showed that DIG decreased the viability of H460 cells and downregulates phospho-AKT (that indicates an active PI3K/AKT pathway) in cells growing under RCCs. However, in LTs, where H460 cells were cultivated in the absence of external mitogenic stimulation and the PI3K/AKT pathway is largely inactive, DIG (although with lower potency) was still able to decrease cell viability. Conversely, while SOX2 is highly expressed in LTs, it is extremely low in cells growing under RCCs, and its levels are further downregulated by DIG. 12
Regardless of the ultimate mechanism of action for NIG, this study demonstrates that cells displaying different degree of proliferation and stemness can be targeted and eliminated by a combination of NIG and DIG or MD at therapeutic levels and warrants further assessment using both in vitro and in vivo studies.
Conclusion
Our data demonstrate that NIG is able to decrease the viability of cells displaying varying rates of proliferation and degrees of stemness, and that this effect can be potentiated by therapeutic levels of the US Food and Drug Administration (FDA)-approved drugs DIG and its novel synthetic analog MD. In addition, we have identified that NIG downregulates the expression of key proteins of Wnt canonical signaling pathway important for properties associated with stemness maintenance, tumorigenesis, and metastasis such as migration and invasion, but at the same time promotes its translocation into the nucleus. The latter suggests that the effect of NIG on cell viability is the result of the activation of the Wnt pathway.
Footnotes
Acknowledgements
The content is solely the responsibility of the authors and does not represent the official views of the National Institutes of Health.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
This study was supported by grants CA173069 from the National Cancer Institute (NIH/NCI) to A.K.V.I. and HL112630 to N.A.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.