
Abstract
Circular ribonucleic acids are non-coding ribonucleic acids that can be identified from genome sequencing studies. Although they can be readily detected, their regulation and functional role in human diseases such as cancer are unknown. Using a systematic approach, we analyzed ribonucleic acid-sequencing data from a well-characterized cohort of intrahepatic cholangiocarcinoma to identify genetic pathways related to circular ribonucleic acids. Although the expression of most circular ribonucleic acids was similar in both the cancer and non-cancer tissues, expression of circ2174 was significantly increased in cancer tissues. Network analysis of co-related genes identified several pathways associated with circ2174, and common regulatory mediators between genes in these pathways and circ2174. Among these, alterations in several genes involved in interleukin-16 signaling responses such Lck, interleukin-16, and macrophage inflammatory protein-1-beta were the most prominent. Octamer transcription factor (Oct)-2 was identified as a signal transducer that was common to both circ2174 and interleukin-16. Circ2174 has sequence complementarity to miR149 which can target Oct-2. These data suggest a mechanism whereby circ2174 can act as a sponge to regulate the expression of miR149, and thereby modulate Oct-2 and interleukin-16 signaling pathways in cholangiocarcinoma.
Keywords
Background
Intrahepatic cholangiocarcinoma (CCA) is an aggressive malignancy associated with the small bile ducts within the liver.1,2 The clinical presentation of these cancers is non-specific, and diagnosis is often made when the tumors are at an advanced stage. Consequently, the tumors are associated with a poor prognosis. The molecular pathogenesis of these cancers is poorly understood. The role of non-coding ribonucleic acids (ncRNAs) in malignant transformation of cholangiocytes or tumor growth is becoming appreciated.3,4 These RNA does not encode for protein but are biologically functional. Based on their size, ncRNAs are broadly classified into two groups. Long ncRNAs are greater than 200 nucleotides and include long intergenic non-coding RNAs, natural antisense transcripts, transcribed ultraconserved regions, long enhancer ncRNAs, non-coding repeat sequences, and pseudogenes.3,5 Small ncRNAs are less than 200 nucleotides include micro ribonucleic acids (miRNAs), small nucleolar RNAs, P-element induced Wimpy Testis (PIWI)-interacting RNAs, and endogenous small interfering RNAs. 5 Among the small ncRNA, an oncogenic or tumor suppressor role in cancer has been best described for miRNAs.6,7 miRNAs have been implicated in many cellular processes such as proliferation, differentiation, migration, invasion, and apoptosis and can regulate gene expression by binding to 3′-Untranslated Region (UTR) of messenger ribonucleic acids (mRNAs). In contrast, the roles of other small ncRNA remain largely unexplored.
The circular ribonucleic acid (circRNA) is a distinctive class of endogenous small ncRNA. They are unique in forming a covalently looped structure, unlike linear mRNAs. The circRNAs can be generated from introns and/or exons following non-canonical alternative splicing. CircRNAs have been implicated in disease related pathways and processes such as embryonic development and aging. 4 A role for some circRNAs in cancers is emerging. In CCA cells, the circRNA SRY was reported to inhibit cell migration and invasion. 4 Some circRNAs can act to sequester miRNA, acting as a sponge that prevents miRNA from binding to their target genes. Other mechanisms that have been described for other circRNA include the regulation of transcription through interaction with RNA polymerase II complexes, or providing templates for the initiation of translation, or interactions with proteins to modulate protein function.
As with many other types of non-coding RNA, deciphering the specific biological contributions of circRNAs is necessary in order to understand their roles in disease pathobiology. Using a network-based gene transcriptomic analysis, we sought to obtain insights into intermediate regulatory elements between circRNA genes, their potential regulators, and biological mechanisms in CCA. We performed a weighted gene co-expression network analysis (WGCNA) analysis using RNA sequencing data from a well-annotated and characterized cohort of intrahepatic CCA. This analysis presumes that strongly correlated expression levels of a group of genes indicate cooperativity among those genes within related pathways, and that these contribute to the resulting phenotype. In addition, genes may also cluster together as a result of a common set of transcription factors, the identity of which can also be detected by systems analysis. Although circRNAs are stable and ubiquitous, characterization of their contribution and involvement in the regulation of gene expression are challenging. This systemic approach provided insights into the contributory roles of circRNA to gene expression in CCA. This approach can be extended to other cancers or diseases and generate new hypotheses regarding the biological roles and functions of circRNAs.
Methods
Tumor cells and tissues
Data were based on analysis of RNA sequencing data from 36 patients with intrahepatic CCA in the National Cancer Institute’s The Cancer Genome Atlas (TCGA) database. Human intrahepatic CCA cell lines CCLP1 and SG231 were obtained and cultured as previously described.5,8,9
RNA sequencing analysis and quality control
Raw reads from RNA sequencing from 36 intrahepatic CCA were downloaded from the TCGA CGHub as FASTQ files. All files were then processed using the exceRpt small RNA-seq pipeline version 4.6.2 (ref). Reads were first aligned to UniVec contaminants and ribosome RNAs using Bowtie aligner. 10 Non-mapped reads were then aligned using STAR to human genome build hg19, Gencode genes, and circular RNAs sequences in circBase 11 in parallel. Subsequently, raw read counts were obtained for circular RNAs and RNAs defined in Gencode, which were further normalized to reads per million (RPM). For quality control, principal component analysis was performed on the normalized gene expression profile using R function prcomp to identify any outliers. Coding RNAs defined in Gencode were selected for further analysis based on annotations as either “protein_coding” (contains an open reading frame) or “ambiguous_orf” (believed to be protein coding, but with more than one possible open reading frame). Coding RNAs with low expression (median log2(RPM) was less than –1) were removed.
Bioinformatics and database analyses
Gene co-expression analysis was performed using R package WGCNA. 12 This approach is based on correlation between expression profiles to detect sets of highly co-regulated genes that can reveal complex transcriptional regulation mechanisms. Clusters of co-regulated genes that share common function “modules” are thought to work together in a network that may correspond to biological pathways. This analysis presumes that strongly correlated expression levels of a group of genes will identify those genes that work cooperatively in related pathways and contribute to the resulting phenotype. In addition, genes may also cluster together as a result of a common set of transcription factors. TargetScan was used for the prediction of miRNA targets.13–16 Interactions between circular RNA and miRNA were analyzed used Circinteractome, which was also used to design divergent circRNA primers as well as siRNAs directed against circRNAs. 17 cBioportal was used to determine the correlation between genes in TCGA intrahepatic CCA datasets. Expression of selected proteins in CCA tissues was examined using the Human Protein Atlas.16,18
Correlation analysis
For each of the top five expressed selected circRNAs, Spearman correlation was calculated of its expression and coding RNA expression in 35 samples using R function cor. For each of the selected circRNAs, the top 50 positively correlated coding RNAs and top 50 negatively correlated coding RNAs were selected for pathway and functional enrichment analysis.
Functional enrichment analysis
Pathway and gene ontology (GO) enrichment analyses were performed using MetaCore v6.31. The background gene list composed of all expressed coding RNAs identified as indicated above. Enrichment analyses were performed using the groups of genes that composed of the top 50 positively or negatively correlated coding mRNAs with each of the five circRNA of interest.
Module network inference
We used the LemonTree software package that incorporates state-of-the-art ensemble methods for module network inference 19 as an established statistical method to reconstruct co-expression modules and their upstream regulatory programs from integrated multi-omics datasets. A set of transcriptional regulators was identified using the GO terms for “transcription factor activity” and “signal transducer activity.” These were then applied to nodes and the corresponding probabilistic scores for being a regulator were then determined. 19 The read counts (RPM) of coding gene and circular RNA were log2 transformed and filtered so that the genes with median transformed expression <–1 were excluded. The expression matrix was then standardized across samples so that each gene or circular RNA had a mean value of 0 and a standard deviation of 1. From the standardized expression matrix, the GaneSh subroutine (in Java) was used to identify co-expression gene modules. 20
siRNA transfection
siRNA against circ002174 was purchased from Dharmacon (Lafayette, CO). For siRNA transfection, SG231 cells were plated in six-well plates at seeding density of 4 × 105 cells in 2 mL complete media. Lipid mediated delivery of siRNA was carried out using Lipofectamine 2000 (Invitrogen, Carlsbad, CA) in antibiotic free media. After 6 h, the media was replaced with complete media and cells were incubated for 48 h.
RNA isolation and quantitative polymerase chain reaction
Total RNA was isolated from the cells using TRIzol reagent (Invitrogen). The expression of circ002174 and Oct-2 was assessed by first generating cDNA using the Biorad iScript cDNA synthesis kit (Hercules, CA), followed by quantitative reverse transcription (qRT)-polymerase chain reaction (PCR) using SYBR green (Takara, Kyoto, Japan) and a Light Cycler 96 System (Roche Diagnostics, Mannheim, Germany). The expression of miR149 was assessed by first generating cDNA using the Taqman micro RNA Reverse transcription kit (Applied Biosciences, Foster City, CA) followed by qRT-PCR using Taqman Universal PCR Master mix (Applied Biosciences).
Results
Identification of circular RNA from patients with CCA
The patient cohort comprised of 36 patients with intrahepatic CCA. Figure 1 provides clinical and demographic information about this cohort. RNA sequencing data obtained from tumors from these patients was re-analyzed using a standardized bioinformatics analysis pipeline (Figure 2). Coding and circular RNA were identified from alignment and annotation of read count data using Gencode and circBase databases. The number of mapped reads to genes in Gencode ranged from ∼28 to ∼73 million with an average of ∼50 million and were normalized to RPM. One outlier (TCGA-W5-AA2H-01A-31R-A41I-07) was detected by principle component analysis and was excluded from downstream analysis (Supplemental Figure 1A). Next, we removed coding RNAs with low expression defined by a median log2(RPM) < –1 and retained 14097 coding RNAs (Supplemental Figure 1B). A total of 462 circRNA were identified. However, the expression levels of annotated circRNA were very low. The top 5 circRNA with the highest read counts in human CCA samples are listed in Table 1 are were selected for further study. Among these, the most highly expressed circRNA was circ2174. Circ2174 is an intergenic circular RNA, with a genomic length of 18630, and genomic location that overlaps with the location of the immunoglobulin heavy constant epsilon (IGHE) gene on chromosome 14. The correlation between each of the top five circRNAs is listed in Table 2. A positive correlation was noted for circ2174 and circ2164. Both of these circRNA arise from the same gene region suggesting the presence of a common mechanism of regulation of expression of these circRNA in CCA.
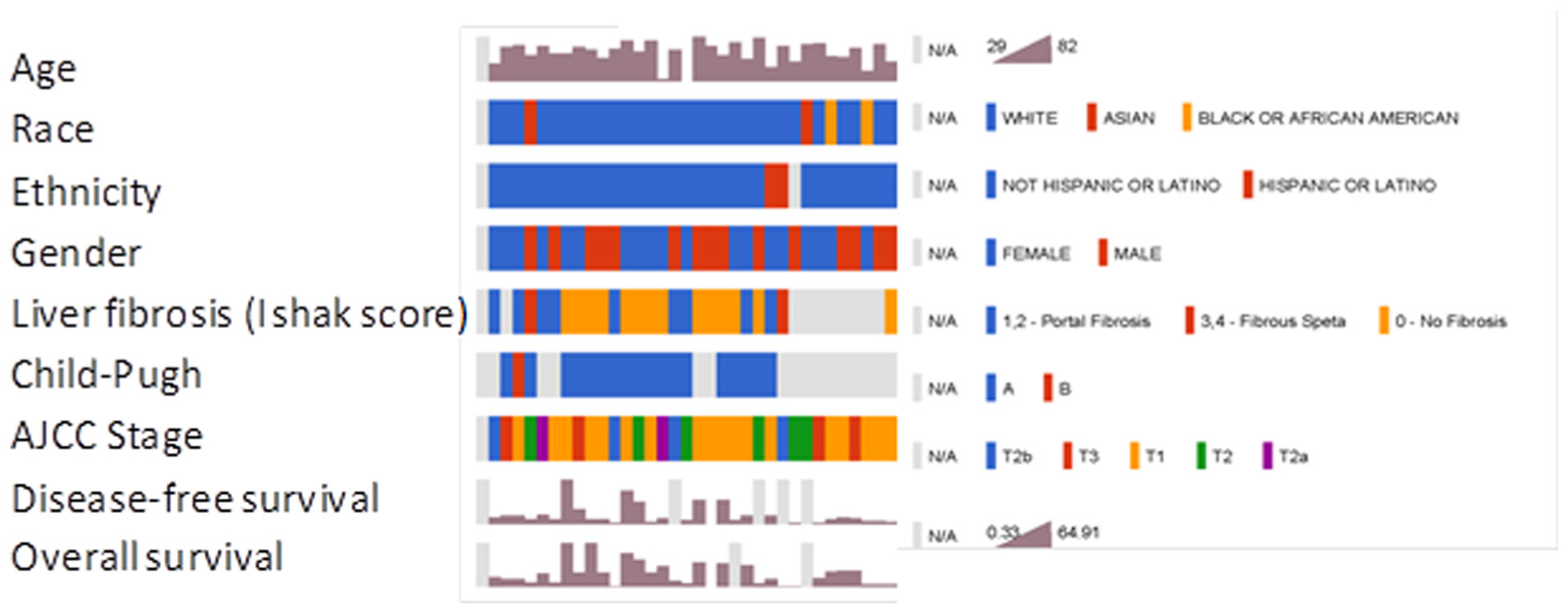
Demographic and clinical characteristics of CCA patients. Each column represents one of 35 patients with CCA included in this study. Demographic information, characteristics of the underlying liver disease, and outcomes are outlined.

Workflow of the Bioinformatics analysis pipeline. A sequential workflow of the steps from the initial input of raw data to analysis of the processed data is shown. The exceRpt Small RNA-seq pipeline was used for all analyses.
Highly expressed circRNA in human CCA samples.

circRNA: circular ribonucleic acid; IGHE: immunoglobulin heavy constant epsilon; IGHG1: immunoglobulin heavy constant gamma 1.
Correlation of circRNAs expressed in intrahepatic cholangiocarcinoma.
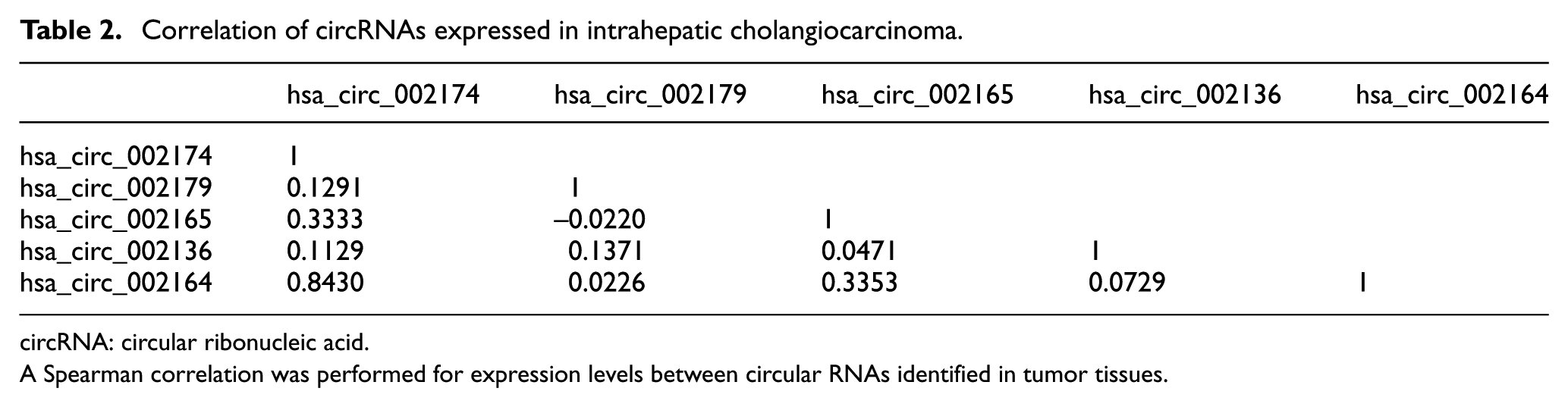
circRNA: circular ribonucleic acid.
A Spearman correlation was performed for expression levels between circular RNAs identified in tumor tissues.
Network analysis
A systematic approach was then used to identify protein coding mRNA that may be correlated with each of these circular RNA. The WGCNA approach was used to analyze the co-expression of circRNA with mRNA. WGCNA first calculated a correlation matrix using expression profile of 14097 coding RNAs plus 462 circRNAs, which was then transformed to a topological overlap matrix (TOM) to better capture hidden gene–gene correlations. Genes were hierarchically clustered based on TOM resulting in 44 clusters of highly co-expressed genes (modules) where genes within the same module had similar expression pattern in 35 CCA samples (Figure 3). Module 0 (M0) was designated as a special module and contained all genes that did not belong to any co-expression modules. All other modules were ranked by the number of genes in it such that module 1 (M1) had the most genes. Forty modules had more than 50 genes. For each co-expression module excluding M0, a single eigengene was calculated to represent the expression pattern of that module. For each of the genes belonging to a co-expression module, correlation with the module eigengene was calculated to reflect the centrality of this gene in the module, which was also designated as module membership. The circRNA were individually correlated with mRNA, and correlation was performed using the Spearman correlation coefficient. p values were adjusted using the Benjamini–Hochberg correction.

Gene dendrogram. Visual representation of co-expression modules identified using weighted gene co-expression network analysis. Each vertical line represents a single gene, and each color bar represents a single module.
Co-expression of mRNA and circRNA was noted only in a single cluster (module 4) which composed of 707 coding RNA and 9 circRNAs. To identify co-expression with miRNA, we separately examined co-expression of circRNA with both mRNA and miRNA. Similar to prior results, a separate cluster was identified which composed of 726 genes. The gene composition of this cluster was very similar to the previously identified module 4 comprising of co-expressed circRNA and mRNA. The additional genes identified were miRNA that are co-regulated with mRNA within this module, confirming the ability of this approach to detect biologically linked genes.
To identify co-expressed circRNA and miRNA, we next specifically analyzed co-expression of circRNA with miRNA and identified one module which contained only a single miRNA, mir650. The expression level this miRNA in our samples was very low. Based on these observations, we concluded that circRNA expression in intrahepatic CCA occurred independently from miRNA, and that the mechanisms of transcriptional regulation of circRNA and miRNA expression were distinct and separate.
Functional enrichment analysis
In order to identify functional correlates of co-expressed genes, we examined GO defined pathways that were over-represented within a selected module. A comparative enrichment analysis was performed comparing possible targets and functional ontologies for genes within the selected module, module 4, with those in the overall gene set using MetaCore. The probability of a random intersection between a set of the size of the module 4 target list with ontology entities was examined. A lower p value of hypergeometric intersection indicates a higher relevance of the entity to the dataset and results in a higher rating. The most significant pathways and GO processes associated with module 4 are summarized in Tables 3 and 4.
Enrichment by process network.

FDR: false discovery rate.
The top 10 networks within module 4 are listed. Each process represents a predefined network of protein interactions characteristic of the process where content is defined manually and annotated.
Enrichment of gene ontology (GO) cellular processes in Module 4.

FDR: false discovery rate.
The top 10 terms are listed. As most GO processes have no gene/protein content, the “empty terms” are excluded from p value calculations.
As this module is large, with >700 genes, we next sought to further refine our analysis to reduce the module membership. Thus, we examined specific co-relationships between genes that were highly correlated with individual circRNA within this module. We identified those genes in module 4 that had a correlation coefficient with circ2174 of 0.75 or more, and with a p value of 10–6 or lower. The top 50 most highly correlated mRNA with circ2174 are listed in Supplemental Table 1. Functional enrichment analysis of these genes revealed several pathways (Table 5). Several of the positively correlated pathways are involved in immune or inflammatory responses. Among these, genes such as Lck, interleukin-16 (IL-16), mature or N-terminal IL-16, and macrophage inflammatory protein-1-beta (MIP-1-beta) that are part of the IL-16 signaling pathway (p value 3.766 × 10–6) were particularly prominent.
Pathway enrichment analysis.

IL-16: interleukin-16; MIP-1-beta: macrophage inflammatory protein-1-beta; FDR: false discovery rate.
Pathways associated and positively correlated with circ2174.
Identification of transcriptional regulators
We next sought to identify common mechanisms of transcriptional regulation. An integrative module network inference analysis was performed using LemonTree and GaneSh to identify potential transcriptional regulators, and 783 co-expression modules were identified. Circ2174 was contained within module 555. Candidate transcriptional regulators of module 555 were then identified using genes with the GO terms “transcription factor activity” (GO:0003700) and “signal transducer activity” (GO:0004871) or their offspring terms. From 848 genes with transcription factor activity and 740 genes with signal transducer activity that were within our expression profile, POU2F2/Oct-2 was identified as the most probable transcriptional regulator of co-expressed genes within module 555. Interestingly, POU2 F2/OCT2 is a common regulator of both circ2174 and IL-16. These observations were validated in intrahepatic CCA tissues. A high co-expression was observed between Oct-2 and several genes involved in IL-16 signaling using cBioportal, with a Pearson’s correlation coefficient of Oct-2 with IL16, Lck, and MIP-1-beta of 1.00, 0.97, and 0.58, respectively.
miRNAs that could potentially target Oct-2 can be identified based on the presence of complementary sites that match the seed region of miRNAs. Interestingly, several of these have binding sites on circ2174. These include miR-149-5p, miR-874-3p, and miR-582-5p. Moreover, for these miRNA, there are multiple binding sites present on circ2174, with miR-149, miR-874, and miR-582-3p having 10, 16, and 2 binding sites, respectively. To further examine the impact of circ2174 expression, siRNA was used to modulate the expression of circ2714 in SG231 CCA cells, and the effects on Oct-2 and miR-149 assessed using PCR. The efficacy of knockdown varied considerably across biological replicates, and thus, a correlation analysis was performed to examine the relationship between circ2174 levels and those of Oct-2 and miR-149 (Figure 4). In SG231 cells, transiently transfected with siRNA to circ2174, there was a concomitant reduction of Oct-2 expression (Figure 5). These observations suggest a model whereby circ2174 could act as a sponge to sequester miRNA, and thereby limits its effects on regulation of expression of target gene such as Oct-2, and subsequently on downstream pathways regulated by Oct-2 (Figure 6).
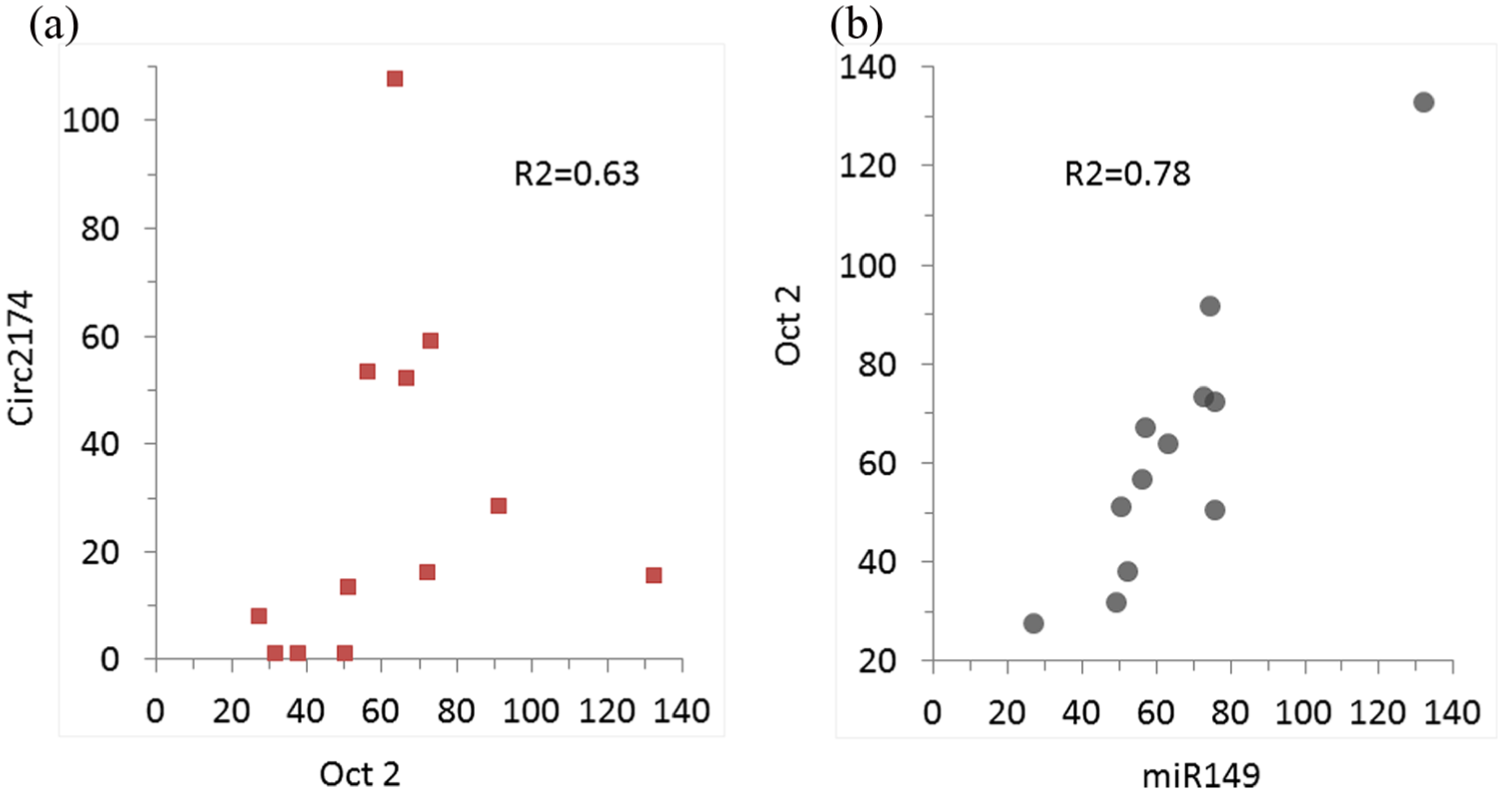
Correlation between Oct-2 and non-coding RNA. RNA expression was determined by qPCR and expressed relative to that of U6 RNA as expression units (2–ΔCT × 106) (a) Correlation between Oct-2 and circ2174. The R2 value was 0.62. (b) Correlation between Oct-2 and miR149. The R2 value was 0.78.

Effect of modulation of circ2174 on Oct-2. SG231 cells were transiently transfected with siRNA to non-targeting control or siRNA to circ2174 which reduced circ2174 RNA expression by almost 50%. After 48 h, RNA was isolated and expression of Oct-2 and U6 determined by qPCR. Data represent mean and standard error of Oct-2/U6 mRNA expression relative to controls from three separate determinations. *p = 0.04.
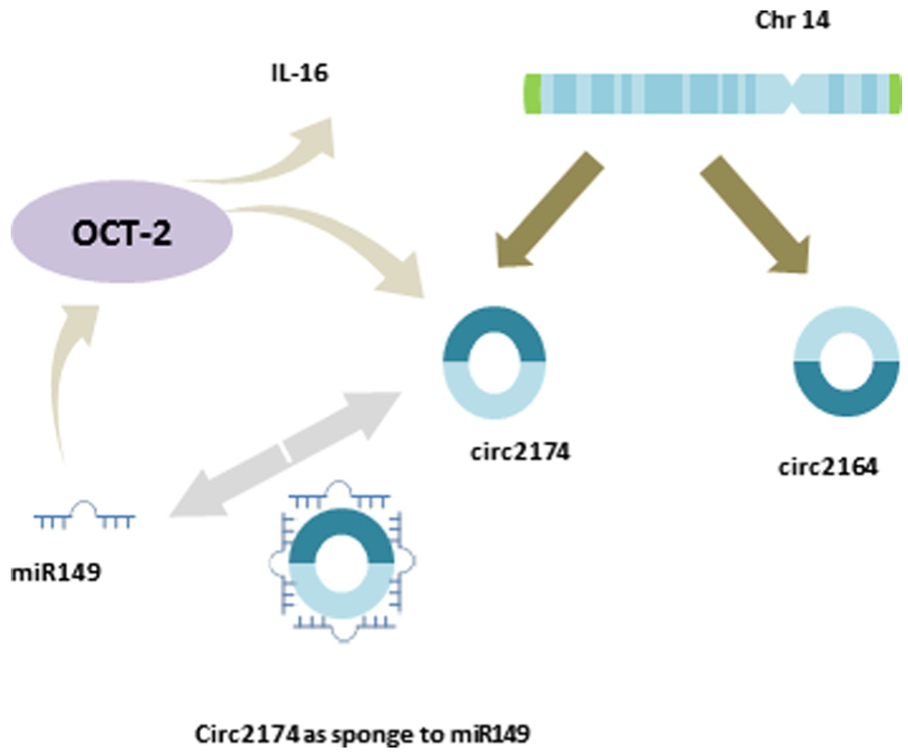
Conceptual model. A hypothetical model is suggested whereby circ2174 can bind to miR-149 and act to sequester this miRNA, thereby modulating the effects of miR-149 on target genes such as Oct-2. The presence of an auto-regulatory loop is suggested by the predicted co-regulation of circ2174 and Oct-2.
Discussion
Using a network-based gene transcriptomic analysis, we sought to obtain insights into intermediate regulatory elements between genes, regulators, and biological mechanisms in intrahepatic CCA. This study demonstrates the utility of a systems biology approach to unravel the network biology and inter-relatedness of non-coding RNA such as circRNA whose functional contributions to network biology may be difficult to decipher. Deregulated expression of circRNA was not a prominent occurrence in CCA, and the expression levels of circRNA were low. Although this study identified only a small number of circRNAs, they are co-expressed with several mRNAs and therefore may actively modulate several genes present in those associations. We identified circ2174 as an intergenic circRNA that is expressed in intrahepatic CCA. The genomic location of Circ2174 is associated with IGHE, a protein coding gene associated with the innate immune system and Interleukin-4 and 13 signaling. However, a lack of correlation between circ2174 and IGHE expression indicated that regulation of expression of these genes can occur independently of each other.
Using systematic analyses of comprehensive RNA sequencing and exome-sequencing data from human CCA tissues, we observed an association of this circRNA with Oct-2, and a correlation between Oct-2 and both circ2174 and miR149. Circ2174 has 10 binding sites for miR149, and moreover, miR149 can target Oct-2 and regulate its expression. Both Oct-2 and miR-149 have been implicated in other cancers. Oct-2 is a transcription factor expressed normally in B cells and B cell lineage tumors regulating B cell proliferation and B cell differentiation genes. 21 In gastric cancer, Oct-2 is highly expressed in metastatic cells while in pancreatic cancers, Oct-2 has been postulated as a bio-marker.21,22 miR149 has been postulated to have a tumor suppressor role in several different cancers to suppress migration, invasion, and metastases of breast cancer, induction of apoptosis in laryngeal or renal cancers or contribute to IL6 activation in gastric cancer.23–28 In hepatocellular carcinoma, miR149 was significantly downregulated in the setting of distant metastasis. 7
The potential for enhanced expression of Oct-2 to contribute to cancer progression in CCA warrants further assessment. These would involve an assessment of the role of Oct-2 on cell growth. In addition, by acting as a sponge to miR-149, circ2174 could decrease the tumor inhibitory effects of miR-149 and further modulate the expression of Oct-2 in an auto-regulatory loop. Circ2174 was noted to be positively correlated with several genes involved in the IL16 signaling pathway. Notably, Oct-2 is a common signal transducer to both circ2174 and IL16, and moreover, IL16 strongly correlates with Oct-2 mRNA in the TCGA intrahepatic CCA RNA-seq dataset. Thus, modulation of Oct-2 expression could impact multiple pathways that may contribute to disease pathobiology.
In conclusion, new insights into the pathogenesis of CCA and other cancers may arise from integrated analyses such as TCGA based on systems biological approaches, and the use of network analyses to decipher function. This network analytical stratagem enables hypothesis generation regarding the transcriptional regulation and provides a first step into understanding new pathways and more importantly, key regulator non-coding genes present in those pathways which give a better understanding of the complex transcriptional regulation of human cancers.
Supplemental Material
Supplemental_Table_and_Figure – Supplemental material for Network analyses–based identification of circular ribonucleic acid–related pathways in intrahepatic cholangiocarcinoma
Supplemental material, Supplemental_Table_and_Figure for Network analyses–based identification of circular ribonucleic acid–related pathways in intrahepatic cholangiocarcinoma by Anuradha Moirangthem, Xue Wang, Irene K Yan and Tushar Patel in Tumor Biology
Footnotes
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the Office of the Director, National Institutes of Health (USA) through award UH3 TR000884.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.