
Abstract
Cholangiocarcinoma is a malignancy of bile duct epithelia with an increasing in incidence rate worldwide. Surgery is the only curative treatment, while adjuvant chemotherapy and radiotherapy render poor responses. Cell-based immunotherapy is a potential strategy for cholangiocarcinoma treatment. However, variation of tumor antigens in cholangiocarcinoma leads to the ineffectiveness of cell-based immunotherapy. In this study, we examined the activation of effector T-cells by dendritic cells pulsed with protein lysate or total RNA from cholangiocarcinoma cell lines for their cytolytic activity against cholangiocarcinoma. Broad-spectrum antigen types with respect to RNA antigen sources were obtained from combination of three cholangiocarcinoma cell lines (KKU-213, KKU-100, and KKU-055). Compared with protein lysate–pulsed dendritic cells, total RNA–pulsed dendritic cells induced anti-tumor effector T-cell response with higher killing ability to KKU-100 and KKU-213 cells compared with protein lysate–pulsed dendritic cells. Moreover, pooled messenger RNA from three cholangiocarcinoma cell lines significantly increased the specific killing capacity of activated lymphocytes against KKU-213 cells. These results suggest that activation of anti-tumor effector T-cells against cholangiocarcinoma by RNA-pulsed dendritic cells is more effective than that by protein lysate–pulsed dendritic cells. In addition, pulsing dendritic cells with pooled messenger RNA from multiple cell lines enhanced the efficacy of a cellular immune response against cholangiocarcinoma.
Keywords
Introduction
Cholangiocarcinoma (CCA), a malignancy of bile duct epithelia is an important cancer in the northeast of Thailand, 1 and its incidence and mortality rates are increasing worldwide.2,3 Epidemiological and experimental studies have shown that risk factors for CCA in Asian populations are different compared with those in the Western populations. 4 Infection by the liver fluke, Opisthorchis viverrini (OV) is the major risk factor for CCA in Asian populations. 5 CCA is a highly infiltrating tumor with high metastasis to lymph node and blood vessel. The disease is normally difficult to diagnose until it becomes advanced or disseminated, at which stage the prognosis is poor. 6 Surgery is the only curative treatment for patients with CCA; however, less than one third of patients are resectable at diagnosis.7,8 The disease is well known to recur after surgery, and there is a high mortality rate even if the patients have received operation. 9 Moreover, the use of adjuvant chemotherapy for CCA renders poor outcomes, and studies examining the effectiveness of treatment are conflicting.10,11 At present, there is no standard protocol for successful treatment for CCA. Therefore, the development of new treatment strategies is required.
Cellular immunotherapy is a promising strategy for cancer treatment, with low toxicity to normal tissues but high capacity to destroy tumors. This strategy involves the use of autologous immune cells, such as dendritic cells (DCs), adoptive T-cells, and engineered chimeric antigen receptor (CAR) T-cells. 12 DCs are potent antigen-presenting cells that control a spectrum of innate and adaptive immune responses and can effectively activate anti-tumor effector cells, for example, cytotoxic T lymphocytes.13,14 DCs can also be used to activate effector T-cells to generate adoptive T-cells 15 potential for cancer treatment. An important step in generation of adoptive T-cells by DC activation is the selection of specific antigens for pulsing DCs to maximize the anti-tumor immune response. The most commonly used antigen type is a peptide from specific antigens. 16 However, induction of the immune system by a restricted repertoire of T-cell clones limits the response to broad tumor antigen variation. 13 Another approach is loading DCs with whole tumor antigen, such as tumor protein lysate, which can produce large repertoires of T lymphocytes and an improved immune response. Tumor cell lysate has been widely used as whole antigen for pulsing DCs in cancer immunotherapy, including non-OV-associated CCA. 17 However, a limited efficiency of DCs loaded with tumor protein lysate has been reported.18,19
An alternative strategy is loading DCs with tumor RNA. Pulsing DCs with total RNA has been attempted in many studies with increasing success, and it has efficiently stimulated anti-tumor T-cells in clinical trials for renal cell carcinoma, 20 prostate cancer, 21 and melanoma. 22 Established cancer cell lines provide a simple source for RNA preparation to pulse DCs for effective stimulation of the adoptive T-cells or induction of the anti-tumor immune response. Clinically, RNA-pulsed DCs was found to potentially induce favorably activated anti-tumor T-cells for hepatocellular carcinoma (HCC) patients. 23 These studies suggest the potency of tumor RNA antigens extracted from cancer cell lines for induction of anti-tumor immune responses in vitro and in vivo. For CCA, loading of tumor RNA to DCs showed an anti-tumor efficacy in vivo. 24
In addition to the choice of antigen, intra-tumor heterogeneity also causes the difficulty of CCA treatment. Genetic or phenotypic diversity of a tumor results in a mixture of tumor sub-populations within an individual CCA patient. 25 It increases the variation of tumor antigens expressed by this cancer. Such complexity within a tumor contributes to therapeutic resistance and an increased recurrence rate, leading to a high mortality rate.25,26 Use of antigens from one established CCA cell line may not adequately induce polyclonal cytotoxic T-cells against a diverse population of CCA sub-clones. This could permit the remaining unresponsive cells to survive and progress.
A combination of CCA cell lines may create a more diverse mixture of tumor-associated antigens (TAAs), which can broaden the anti-tumor immune response activated by DCs. This combination can therefore provide DCs capable of presenting a wide range of antigens to activate a broader repertoire of cytotoxic T-cells. The multiple clones of effector T-cells generated can then recognize and respond to a wider population of cancer cells, resulting in an increased killing capacity of activated effector immune cells. This study, thus, aims to investigate an antigen type for pulsing DCs that maximizes the anti-tumor immune response against CCA. And, to overcome the problem of intra-tumor heterogeneity, tumor antigens from a number of CCA cell lines were combined for pulsing DCs to improve the efficacy of an anti-tumor immune response against CCA.
Materials and methods
Cell culture
Japanese Collection of Research Bioresources Cell Bank (JCRB1568), KKU-213 (JCRB1557), KKU-055 (JCRB1551)—and an immortalized cholangiocyte cell line—MMNK-1 (JCRB1554) were obtained from the Japanese Collection of Research Bioresources Cell Bank (JCRB), National Institute of Biomedical Innovation, Japan. KKU-100 is a poorly differentiated CCA cell line established from the 65-year-old patient with CCA, 27 and KKU-213 is a mixed (papillary and non-papillary) CCA cell line established from the 58-year-old patient with CCA. Both were opisthorchiasis-associated CCA established and characterized at the Liver Fluke and Cholangiocarcinoma Research Center, Khon Kaen University, Khon Kaen, Thailand. These cell lines were deposited to JCRB for long-term storage and making available as research bioresources. Cell lines were maintained in Dulbecco’s Modified Eagle’s Medium/Nutrient Mixture F-12 (DMEM/F-12; Gibco, Auckland, New Zealand), containing 10% of fetal bovine serum (FBS; Gibco), 100 units/mL of penicillin, and 100 μg/mL of streptomycin at 37°C with 5% CO2 and sub-cultured twice a week by following a standard trypsinization protocol.
Preparation of protein lysate and RNA isolation
Confluent cultures of KKU-100 cells were detached, washed twice in phosphate-buffered saline (PBS), and resuspended in AIM-V medium. The cell suspensions were lysed by rapid freezing using liquid nitrogen and thawing at 37°C for four freeze–thaw cycles. Crude debris was removed by centrifugation. The supernatant was collected and protein concentration was determined by a commercial assay (Bio-Rad, Hercules, CA, USA).
Three CCA cell lines (KKU-100, KKU-055, and KKU-213) were used as antigen sources for DC pulsing. Total RNAs were extracted from CCA cell lines using TRIzol reagent (Invitrogen, Auckland, New Zealand) and chloroform extraction by following the manufacturer’s protocol. Briefly, the cell lines were mixed with 1 mL of TRIzol reagent and 0.2 mL of chloroform and centrifuged. The aqueous phase of the samples was collected and RNA was precipitated with isopropanol and 70% ethanol, respectively. For messenger RNA (mRNA) isolation, the mRNA was isolated from total RNA using GenEluteTM mRNA isolation kit (Sigma-Aldrich, St. Louis, MO, USA) by following the manufacturer’s protocol.
Preparation of peripheral blood mononuclear cells and DC culture
Peripheral blood mononuclear cells (PBMCs) from healthy donors were isolated by Ficoll–Hypaque (GE Healthcare, Freiburg, Germany) gradient centrifugation. PBMCs were subsequently resuspended in AIM-V (Gibco) at 107 cells in a six-well plate and allowed to adhere for 2 h. The non-adherent cells were gently removed and cryopreserved as a source of immune cells. The adherent cells were cultured in AIM-V supplemented with granulocyte–macrophage colony-stimulating factor (GM-CSF; 50 ng/mL; Invitrogen, Frederick, MD, USA) and interleukin 4 (IL-4; 25 ng/mL; Life Technologies, Auckland, New Zealand) to generate DCs. The medium was changed by replacing with fresh medium with the same concentration of cytokines every other day for 6 days.
To generate the protein lysate–pulsed or total RNA–pulsed DCs, DCs were incubated (pulsed) with 60 μg of protein lysate or 10 μg of total RNA from KKU-100 in AIM-V, supplemented with tumor necrosis factor alpha (TNF-α; 50 ng/mL; R&D Systems, Minneapolis, MN, USA) and interferon gamma (IFN-γ; 50 ng/mL; R&D Systems) for 48 h. DCs which were incubated with TNF-α and IFN-γ without the protein lysate or total RNA were used as control DCs.
To generate the mRNA-pulsed DCs, the immature DCs were transfected with mRNA at various conditions using DOTAP (chemical name of N-[1-(2,3-Dioleoyloxy)propyl]-N,N,N-trimethylammonium methylsulfate) liposomal transfection reagent (Roche, Penzberg, Germany) for 5 h. The conditions ranged from RNA of one CCA cell line (1 µg of mRNA from KKU-213 cell line), two CCA cell lines (1 µg of mRNA from KKU-213 and KKU-100 cell lines or 1 µg of KKU-055 cell line), and three CCA cell lines (1 µg of mRNA from KKU-213, KKU-100, and KKU-055 cell lines).
Generation of mature DCs
Immature DCs after incubation with tumor antigens or transfection with mRNA were cultured in AIM-V supplemented with TNF-α (50 ng/mL; R&D Systems) and IFN-γ (50 ng/mL; R&D Systems) for 48 h. DCs which were incubated with TNF-α and IFN-γ without the protein lysate or total RNA or without mRNA transfection were used as control DCs (unpulsed DCs). These cells were analyzed for DC maturation, and these mature DCs were used for activation of autologous non-adherent cells (lymphocytes).
Effector immune cell activation
To activate effector immune cells, autologous non-adherent PBMCs were added to the mRNA-pulsed DCs at a ratio of 10:1 in AIM-V with 5% human AB serum (Life Technologies, Brown Deer, WI, USA) for 3 days. After that, the non-adherent cells were removed to culture in a new six-well plate in AIM-V medium containing IL-2 (20 ng/mL), IL-7 (10 ng/mL), and IL-15 (20 ng/mL; all from R&D Systems) for 7–10 days with media replenishment for every 2 days.
Flow cytometric analysis
Flow cytometry was performed using the following antibodies: anti-human CD14 FITC and anti-human CD11c PE-Cyanine7 (both from eBioscience, San Diego, CA, USA) antibodies to determine the total DC yield; anti-human CD11c APC, anti-human HLA-DR FITC, anti-human CD40 FITC (all of them from ImmunoTools, Friesoythe, Germany), anti-human CD83 PE, anti-human CD86 (B7-2) PE (both from eBioscience) antibodies to monitor DC maturation and co-stimulatory molecules; and anti-human CD8 PE (eBioscience) to assess cytotoxic T-cell yield. After washing with PBS containing 2% FBS, the cells were fixed with 1% formaldehyde in 2% FBS/PBS. To determine cell apoptosis, cells were stained with annexin V–APC (antigen-presenting cell) and propidium iodide (PI; both from ImmunoTools). Samples were analyzed on FACSCalibur using CellQuest software (BD Biosciences, San Jose, CA, USA).
Cell viability assay
The cytolytic activity of immune effector cells was measured by PrestoBlueTM Cell Viability Reagent (Invitrogen, Auckland, New Zealand) according to the manufacture’s protocol. Briefly, 5 × 103 target cells (KKU-100 or KKU-213) were plated on a 96-well plate. Activated effector immune cells were added at effector:target (E:T) ratios of 5:1, 10:1, and 20:1. After 48 h, effector immune cells were removed and target cells were washed twice. Then, PrestoBlueTM Cell Viability Reagent was added and cells were incubated at 37°C until the reagent color changed. Metabolically active cells were stained. Absorbance was read at excitation of 570 nm and emission of 600 nm as a reference wavelength for normalization, and the percentage of cell survival was calculated. Data are reported as the percentage of number of cell death in comparison to target cells without adding effector cells (set as 0%).
Generation of enhanced yellow fluorescent protein–overexpressed KKU-213 cell
Human embryonic kidney cells (HEK293T cells) were seeded at 2 × 105 cells per 60-mm cell culture dish 24 h before transfection. All plasmid DNA were extracted and purified using the QIAGEN midipreparation kit (QIAGEN, Hilden, Germany). Lentiviral vector carrying enhanced nuclear form of enhanced yellow fluorescent protein (EYFPnuc) was described previously. HEK293T cells were transfected with 2 µg of pMD2.G (vesicular stomatitis virus glycoprotein (VSVG) expression plasmid), 7 µg of psPAX2 (Gag/Pol expression plasmid), and pTripZ-EYFP transfer plasmid using calcium phosphate precipitation method. The viral supernatant was harvested at 48 h post-transfection, filtered through 0.45 µm filter membrane, and transduced into CCA cell line, KKU-213, in the presence of polybrene (8 µg/mL; Sigma-Aldrich). Stable cells overexpressing EYFPnuc were selected using puromycin antibiotic drug (1 µg/mL; Gibco). To induce EYFP expression, the cells were added with doxycycline (1 µg/mL; Bio Basic Inc., Markham Ontario, Canada) before performing a cytolytic assay.
Fluorescence microscopic analysis
Overall, 5 × 104 target cells (EYFP-overexpressed KKU-213) were plated on a 24-well plate. Activated effector immune cells were added at an effector:target (E:T) ratio of 10:1. Cells were co-incubated at 37°C for 24 h. After co-incubation, the cell suspension was removed and replaced with a fresh RPMI-1640 medium. The remaining adherent cells were observed under fluorescence microscope.
Cytolytic activity assay
In total, 5 × 104 target cells (KKU-213 or MMNK-1) were plated on a 24-well plate. Activated effector immune cells were added at an E:T ratios of 2.5:1, 5:1, and 10:1 for 24 h. After co-culturing, the cells were washed twice with PBS containing 2% FBS. Subsequently, the cells were stained with annexin V–APC and PI and analyzed for CCA apoptosis by flow cytometry.
Statistical analysis
Mean and the standard error of the mean from at least three independent experiments were calculated. One-way analysis of variance (ANOVA) and Tukey’s multiple comparisons post-test were used for the analysis of data using PRISM GraphPad, version 5.0 (GraphPad Software, v.5.0, San Diego, CA, USA). Student’s t test was employed to assess the statistical significance of differences between a pair of data sets. p value of <0.05 was considered to be statistically significant.
Results
Comparison of protein lysate and RNA antigen sources from the KKU-100 cell line on DC phenotype
Adherent monocytes were cultured and induced in AIM-V medium supplemented with GM-CSF and IL-4. After 5 days, monocytes were differentiated into immature DCs. Cell phenotypic analysis using flow cytometry showed a significant increase in DC marker (CD11c) expression and a strong decrease in monocyte marker (CD14) expression on the cultured adherent cells (Figure 1(a) and (b)). The adherent cell morphology also changed from a spherical shape to a larger dendritic shape (Figure 1(c)-A and 1(c)-B). This data indicated that immature monocyte-derived DCs were successfully generated in vitro from healthy individual’s blood. Immature DCs were pulsed with protein lysate or total RNA extracted from KKU-100 cells in AIM-V medium supplemented with TNF-α and IFN-γ. Compared with immature DCs (Figure 1(c)-B), the morphology of mature DCs (Figure 1(c)-C) showed an irregular shape, growth in clustered colonies, and development of a significantly larger size and further showed roughness, rigidness, and ruffles on the cell surface that involved protrusions of dendrites.
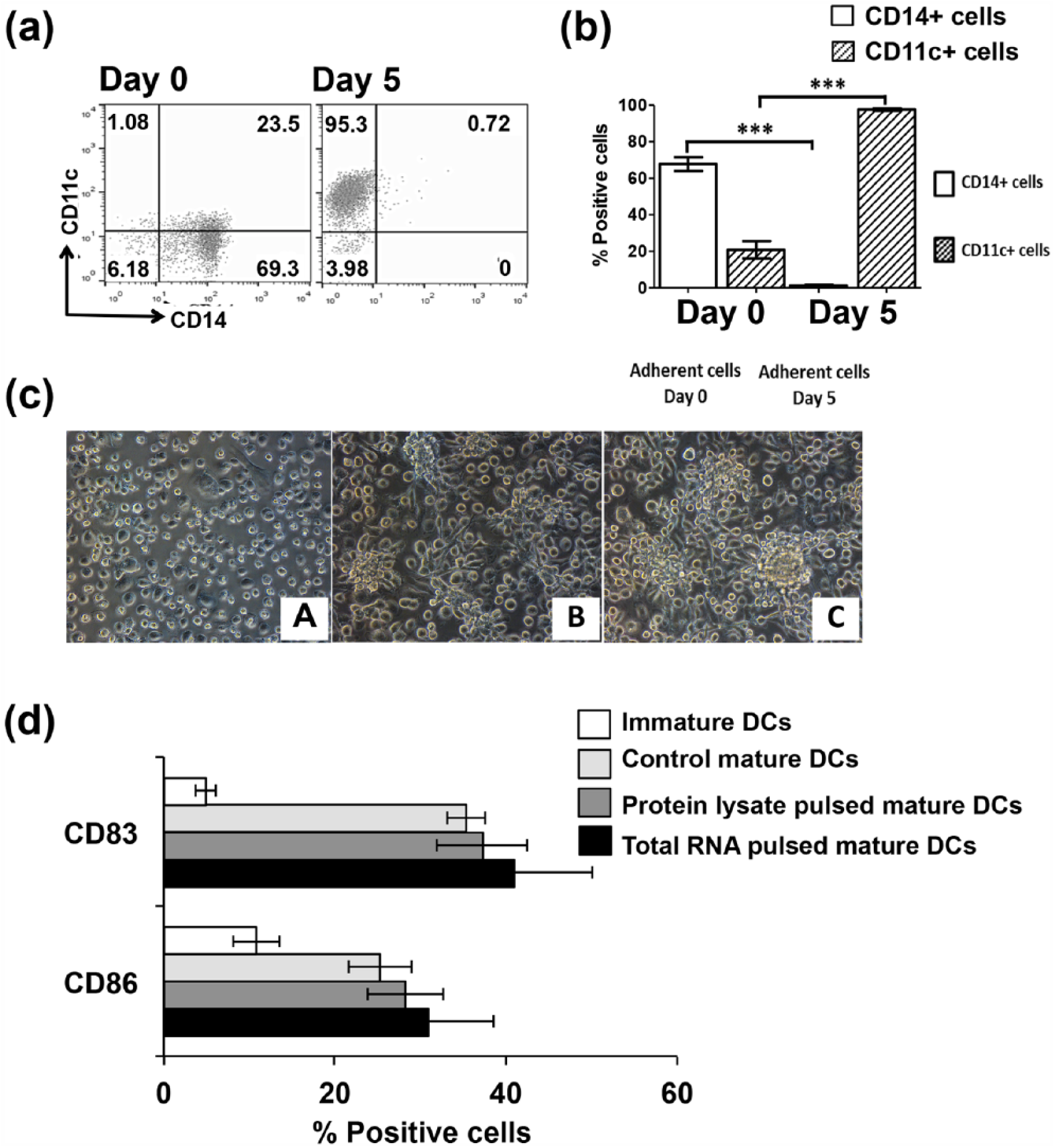
Morphology and phenotype of monocyte-derived dendritic cells. Adherent peripheral mononuclear cells (PBMCs) were cultured in AIM-V medium supplemented with GM-CSF and IL-4. After antigen loading, the immature DCs were then cultured in AIM-V medium supplemented with TNF-α and IFN-γ for 48 h. (a) Expression of monocyte marker (CD14) and DC marker (CD11c) on adherent cells before and after culture in the medium for 5 days, determined using flow cytometry. (b) The percentage of adherent cells expressing monocyte and DC markers. Bar graphs are shown as mean ± standard error of the mean (SEM) calculated from three independent experiments. (c) Cell morphology of (A) monocyte, (B) immature DCs, and (C) mature DCs, observed under light microscope (10× magnification). (d) Surface markers related to DC maturation (CD83) and T-cell co-stimulatory molecule (CD86) were analyzed after antigen pulsing by flow cytometry (***p < 0.001, analyzed by Student’s t test).
After pulsing DCs with tumor protein lysate or total RNA, phenotypic markers of DCs were examined by flow cytometry. DCs with CD11c positive expression were subsequently determined for expression of cell surface markers for maturation status (CD83) and T-cell co-stimulatory molecule (CD86; Figure 1(d)). Immature DCs expressed a low level of CD83 (4.88% ± 1.2%) and CD86 (10.87% ± 2.7%). In control DCs, the expression of CD83 and CD86 also increased (37.25% ± 5.3% and 28.32% ± 4.4%, respectively). However, DCs that were pulsed with CCA protein lysate showed a similar increase in the expression of CD83 (37.25% ± 5.3%) and CD86 (28.32% ± 4.4%). DCs that were pulsed with total RNA showed an increased expression of CD83 (40.68% ± 9.2%) and CD86 (30.87% ± 7.6%). These results indicate that immature DCs were activated to mature DCs and that CCA protein lysate or total RNA increased the expression of maturation markers and T-cell co-stimulatory molecules of the mature DCs.
Comparison of protein lysate and RNA antigen sources from the KKU-100 cell line on effector immune cell populations
To activate effector immune cells, the pulsed DCs were co-cultured with non-adherent PBMCs in AIM-V medium containing IL-2, IL-7, and IL-15. The clonal proliferation of non-adherent PBMCs was observed. After 10 days of culture, the activation markers for effector immune cells were assessed by flow cytometry. The proportion of cytotoxic T lymphocytes, CD3+CD8+ cells, is presented in Figure 2(a) and (b). Compared to the control DCs, protein lysate–pulsed DCs and RNA-pulsed DCs induced higher numbers of CD3+CD8+ cells (with percentages of CD3+CD8+ cells of 31.79% ± 3.73%, 43.37% ± 4.0%, and 62.03% ± 3.9%, respectively). These results demonstrated that the RNA-pulsed DCs induced significantly higher numbers of CD3+CD8+ cells than that of the protein lysate–pulsed DCs. These results indicated that CCA RNA–pulsed DCs increased the number of cytotoxic T-cells more efficiently when compared to protein lysate–pulsed DCs.

Expression of CD8+ T-cells activated with protein lysate–pulsed DCs or total RNA–pulsed DCs. After pulsing DCs with protein lysate or total RNA from CCA cell line (KKU-100), pulsed DCs were then co-cultured with autologous non-adherent PBMCs for 10 days in the presence of IL-2, IL-7, and IL-15. The effector immune cells were collected and expression of CD8+ in CD3+ cells was examined using flow cytometry. (a) Representative dot plots of the data from three independent experiments. (b) Results calculated from three independent experiments are shown as bar graphs, showing percentages (mean ± SEM) of double-positive (CD3+CD8+) cells (*p < 0.05, analyzed by one-way ANOVA followed by Tukey’s multiple comparison tests).
Cytolytic activity of effector immune cells against CCA
Cytotoxic activity of effector immune cells induced by DCs loaded with protein lysate or RNA from KKU-100 cells was determined. Target CCA cells (KKU-100) were co-cultured with effector immune cells for 48 h, and the percentage of KKU-100 cell death was observed using PrestoBlue dye assay. As shown in Figure 3(a), both protein lysate–pulsed and total RNA–pulsed DCs stimulated effector immune cells that recognized and lysed KKU-100 cells. Loading DCs with protein lysate significantly induced cytolytic activity of effector immune cells at effector:target (E:T) ratios of 10:1 and 20:1 (percentages of target cell death were 37.17% ± 3.8% and 53.40% ± 6.23%, respectively). RNA-pulsed DCs induced cytolytic activity of effector immune cells at E:T ratios of 5:1, 10:1, and 20:1 (percentages of target cell death were 37.50% ± 4.32%, 69.60% ± 6.84%, and 78.17% ± 8.35%, respectively). Unlike DCs loaded with protein lysate, RNA-pulsed DCs induced significant cytolytic activity of effector immune cells at an E:T ratio of 5:1. This indicated that loading DCs with total RNA from KKU-100 stimulated greater cytolytic activity of effector immune cells against CCA.
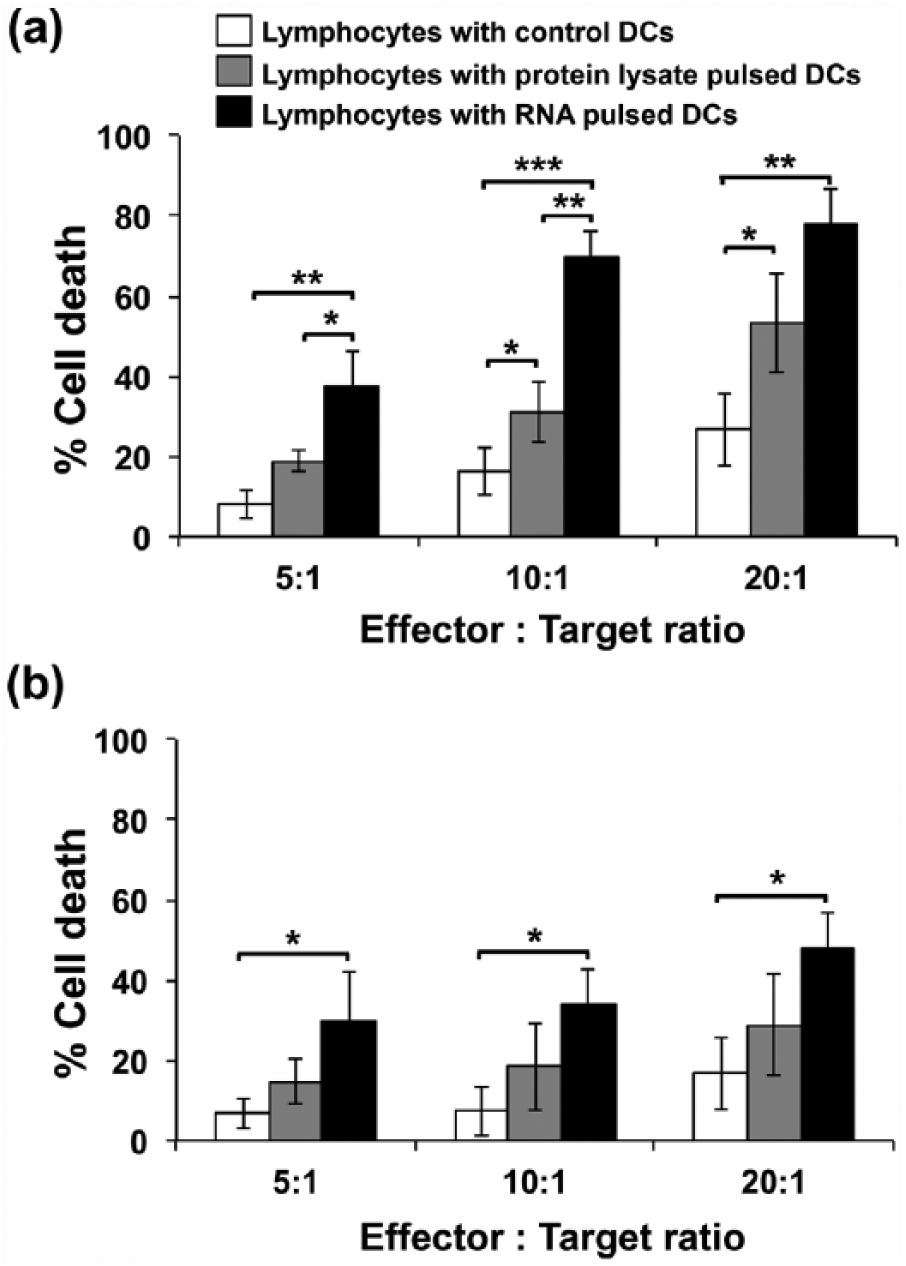
The percentages of target CCA cell death after co-culturing with activated lymphocytes. CCA cell lines were co-cultured with autologous non-adherent PBMCs or lymphocytes (effector cells) that were activated by control DCs (white bar), protein lysate–pulsed DCs (gray bar), or total RNA–pulsed DCs (black bar) for 48 h. The percentage of CCA cell death was determined using PrestoBlue assay. (a) The percentages of cell death of a target CCA cell (KKU-100) at effector:target (E:T) ratio of 5:1, 10:1, and 20:1. (b) The percentages of cell death of a non-target CCA cell line (KKU-213) were also co-cultured to the effector cells with the same E:T ratios. Data represent mean ± SEM of triplicate cultures from three independent experiments (*p < 0.05, **p < 0.01, analyzed by one-way ANOVA followed by Tukey’s multiple comparison tests).
To observe anti-tumor specificity of effector immune cells, cytolytic activity against the non-target CCA cell line (KKU-213) was investigated. Cytolytic activity of effector immune cells that were induced by DCs loaded with protein lysate from KKU-100 cells was not significantly different from that of the control (unpulsed DCs) against KKU-213 cells. However, DCs loaded with RNA from KKU-100 cells induced effector immune cells to lyse KKU-213 cells at E:T ratios of 10:1 and 20:1 with percentages of cell death of 33.93% ± 4.25% and 48.23% ± 4.27%, respectively (Figure 3(b)). This result indicated that DC pulsing with RNA rather than with protein lysate stimulated cytolytic activity of effector immune cells against a different CCA cell line. Due to a higher efficacy for induction of an anti-CCA immune response and the potential for enhanced cytolytic activity against different CCA cell lines, only RNA antigen types were used in combinations of antigens for further experiments targeting intra-tumor heterogeneity.
Effect of pooled mRNA transfection on DC maturation
To enhance anti-CCA responses, mRNAs from three CCA cell lines (KKU-213, KKU-100, and KKU-055) were combined to increase the diversity of CCA antigens for pulsing DCs. In addition, mRNA from KKU-213 was transfected alone or in combination with either KKU-100 or KKU-055, whereas DCs with no mRNA transfection served as control DCs (unpulsed DCs). The effects of pooled mRNA on DC maturation (mDCs) and expression of co-stimulatory molecules were investigated.
Upon 2 days post-transfection with TNF-α and IFN-γ cytokines (day 7 of DC culture), DCs showed increased expression of DC maturation markers (CD83 and HLA-DR (human leukocyte antigen–antigen D related)) when compared to day 5 just prior to mRNA transfection, as shown in Figure 4. However, the increased expression of these markers was similar among the investigated groups. Expression of T-cell co-stimulatory molecules, including CD86 and CD 40, was observed in Figure 4(a) and (b). The results showed a similar percentage of co-stimulatory molecule expression on DCs in all conditions. These data suggested that loading of DCs with KKU-213 mRNA or in combination with mRNAs from one or two distinct CCA cell lines did not affect their maturation.

Expression of maturation markers and co-stimulatory molecules on mRNA-transfected DCs. DCs were transfected with mRNA in various conditions; DCs with no mRNA transfection served as control DCs (unpulsed DCs). After induction with TNF-α and IFN-γ, DCs with or without mRNA transfection were stained with indicated antibodies, followed by flow cytometry. (a) Data represent the percentages and mean fluorescence intensity (MFI) of DC maturation markers, CD83, and HLA-DR and expression of T-cell co-stimulatory molecules, CD86+, and CD40. Black color indicates isotype control. Black line indicates DC maturation or co-stimulatory markers. (b) Results calculated from three independent experiments were plotted as bar graphs, showing percentages (mean ± SEM) of double-positive cells, as indicated (*p < 0.05, **p < 0.01, ***p < 0.001, compared with immature DCs. Data were analyzed by one-way ANOVA followed by Tukey’s multiple comparison tests.).
Apoptotic induction by effector immune cells activated from pooled mRNA–loaded DCs
DCs pulsed with individual or combined mRNA were co-incubated with autologous non-adherent PBMCs for effector immune cell generation. Cytolytic activity of immune effectors was examined to determine whether DCs pulsed from mRNA pools were favorable to enhance an anti-CCA response. The activated effector cells were co-cultured with EYFP-overexpressed KKU-213 cells at a ratio of 10:1 for 24 h. Subsequently, cell suspension containing activated effector cells was removed and the remaining EYFP-overexpressed cells were observed. The results revealed that loading DCs with mRNA pools activated effector immune cells for KKU-213 killing within 24 h, better than those pulsed with mRNA from KKU-213 alone (Figure 5(a)).

Cytolytic activity of the activated effector immune cells against cholangiocarcinoma and non-cancer cholangiocyte cell lines. (a) EYFP-overexpressed KKU-213 cells (target cells) were co-cultured with autologous non-adherent PBMCs or lymphocytes (effector cells) activated by pulsed DCs with mRNA from CCA cell lines as indicated at E:T ratio of 10:1. After 24 h, cell suspensions were removed and the remaining adherent target cells were observed under fluorescence microscope. (A) EYFP-overexpressed KKU-213 cells without effector cells. Remaining EYFP-expressing cells were shown after co-culturing with (B) unpulsed DCs, (C) M213 pulsed DCs, (D) KKU-100+KKU-213 pulsed DCs, (E) KKU-055+KKU-213 pulsed DCs, and (F) KKU-100+KKU-055+KKU-213 pulsed DCs (10× magnification). (b) Target CCA cells (KKU-213) were co-cultured with the effector cells that were activated by DC pulsing from the indicated condition of tumor mRNA at E:T ratios of 2.5:1, 5:1, and 10:1 for 24 h. Cells were collected and stained with annexin V and PI to determine cell apoptosis using flow cytometry. Percentages of cell apoptosis were calculated from three independent experiments. (c) The percentage of immortal cholangiocytes (MMNK-1) apoptosis after 24 h co-culturing with the effector cells. Data represents mean ± SEM of the percentage of cell apoptosis from three independent experiments (*p < 0.05, **p < 0.01, ***p < 0.001, analyzed by one-way ANOVA followed by Tukey’s multiple comparison tests).
The killing capacity of effector immune cells against the KKU-213 cell line was further determined by varying the E:T ratio. The activated effector cells were co-cultured with target cells (KKU-213 cells) at E:T ratios of 2.5:1, 5:1, and 10:1. The percentages of KKU-213 cell apoptosis were determined at 24 h post co-culturing using annexin V/PI apoptosis assay. The percentages of KKU-213 cells apoptosis were increased in a dose-dependent manner. Using mRNA from KKU-213 cells alone in DC pulsing did not efficiently induce cytolytic activity of effector cells against CCA cell lines (Figure 5(b)).
In Figure 5(b), the combination of mRNA from KKU-100 and KKU-213 significantly induced CCA apoptosis at E:T of 10:1. Tumor mRNA from KKU-055 cells was combined with KKU-213 mRNA for DCs transfection, the killing activity of those effector cells was induced even at a low E:T ratio (2.5:1). This indicated that loading DCs with mRNA from KKU-055 and KKU-213 stimulated higher cytolytic activity of effector immune cells against CCA than those loaded with KKU-100 and KKU-213. Figure 5(a) also showed that combination of two distinct tumor mRNAs (KKU-100 and KKU-055) with the KKU-213 mRNA for DC transfection provided the most effective anti-CCA immune response (with the percentages of target cell apoptosis at E:T of 2.5:1, 5:1, and 10:1 were 26.50% ± 3.80%, 37.08% ± 1.46%, and 46.30% ± 7.23%, respectively). In comparison with effectors activated by DCs pulsing with KKU-213 mRNA alone (with the percentages of CCA apoptosis 13.42% ± 2.27%, 17.28% ± 3.66%, and 21.78% ± 3.74%), DCs pulsed with a combination of mRNA from three antigen sources induced killing activity of effector immune cells by approximately twofold in every E:T ratio. This suggested that pooled mRNA from three cell lines in this study enhance cytolytic activity of DCs against CCA cells.
To observe anti-tumor specificity of effector immune cells, cytolytic activity of these effector immune cells induced by individual and pooled mRNA–pulsed DCs was also investigated for their killing capacity against cholangiocyte cell line (MMNK-1). The results showed that effector immune cells did not significantly increase MMNK-1 cell apoptosis (Figure 5(c)). These results indicated specificity of mRNA-pulsed DCs in stimulating cytolytic activity of effector immune cells against CCA but not against non-cancer cholangiocytes.
Discussion
CCA is a fatal disease with a poor prognosis and a high recurrence rate. Currently, there is no effective treatment for CCA. Conventional therapies including chemotherapy and radiotherapy are rather noxious and inefficient. 28 DC-based immunotherapy is an alternative treatment that has been used in clinical trials for many cancers. 15 DC-based immunotherapy has also been studied in non-Opisthorchis viverrini–associated CCA.17,29 In addition, adoptive T-cells activated by DCs pulsed with tumor antigens are potential cellular immunotherapy for CCA. Tumor lysate–pulsed DCs for activated T-cell transfer showed effective treatment by preventing the disease recurrence and prolonging survival. 17 Specific antigen-loading DCs also showed a positive clinical response in CCA patients who had been treated with chemotherapy and showed maintenance of good nutrition status. 29 These studies suggested that the use of effector T-cells activated with pulsed DCs appears promising as an approach for CCA treatment. However, the source of tumor antigens for pulsing DCs required to be explored for the development of adoptive T-cell therapy for CCA. Moreover, their efficacy has also been questioned since intra-tumor heterogeneity increases variations of tumor phenotypes that lead to different responses to the treatment and consequently limiting treatment effectiveness. The remaining unresponsive clones may then overgrow and cause tumor relapse. 30 To overcome this problem, pulsing DC with a mixture of appropriate tumor antigens may increase the probability of generating polyclonal effector immune cells that are capable of simultaneously targeting multiple tumor clones.
In this study, DCs were initially pulsed with protein lysate or RNA from CCA cell lines (KKU-100) that were established from a patient with CCA. DCs were successfully prepared from PBMCs, morphologically appearing as large cells with both round and irregular shapes with many small dendrites (Figure 1(c)). DC maturation is important for antigen presentation to T-cells, which leads to effector cell activation. Therefore, the maturation phenotype of DCs was also observed after loading with tumor antigens. Both protein lysate–pulsed and total RNA–pulsed DCs as well as control DCs expressed the DC maturation marker (CD83) and T-cell co-stimulatory molecular marker (CD86) similarly (Figure 1(d)). This result indicated that loading DCs with CCA protein lysate or total RNA did not affect their maturation and expression of co-stimulatory molecules. In the presence of T-cell co-stimulatory molecules, DCs could induce immune response through T-cell activation. 14 After co-culturing of protein lysate–pulsed or total RNA–pulsed DCs and autologous lymphocytes, activated effector cells were expanded. The effector immune cells that were activated by RNA-pulsed DCs showed higher CD3+CD8+ cell populations compared to effector immune cells that were activated by protein lysate–pulsed DCs (Figure 2). Similar phenomenon was observed in the comparative analysis of HCC cells that pulsing DCs with HCC total RNA showed better cytotoxic T-cell activation than pulsing DCs with HCC protein lysate. 31
The CCA cell lines used in this study were established from Thai CCA patients and the PBMC used for preparation of DCs and lymphocytes were also separated from Thai healthy volunteers. Although HLA compatibility was not tested in this study, the chance for their compatibility was high as HLA-A2, the common type in Thai population. In addition, in the experiments, the control DCs without pulsing was usually included, which showed much lower cytolytic activity of effector T-cells than that of the tests; this might indicate the possibility of their HLA compatibility. The ability of effector immune cells to kill the target CCA cell line (KKU-100) was then examined. However, DCs pulsed with total RNA showed better effector cell activation for target cell killing than DCs pulsed with protein lysate (Figure 3(a)). In addition, the cytolytic activity of effector cells against non-target CCA cell line (KKU-213) was observed but only under conditions of co-culturing with effector cells that were activated by RNA-pulsed DCs (Figure 3(a)). Significant cytolytic activity was observed at E:T ratios of 10:1 and 20:1. The increased proportion of CD8+ T-cells in this study was found to be associated with an increased cytolytic activity. However, natural killer (NK) cells, which were not tested in this study, may also be involved in CCA killing. Roles of DCs on NK cell activation have been reported.32–34 The anti-tumor effect of NK cells after co-culturing with DCs was observed in non–small cell lung cancer, 35 pancreatic carcinoma, 36 and glioblastoma multiform. 37
Since vaccination with DCs is associated with safe and potent effective cell responses, several researchers have evaluated tumor cell lysate and RNA as cost effective sources of antigen loading in various cancers. 38 However, some studies reported that tumor protein lysate limited the immune response by impairing DC function. Tumor cell protein lysate inhibited toll-like receptor (TLR)-induced maturation of DCs in an in vivo model of melanoma. 18 In patients with multiple myeloma, DCs loaded with tumor protein lysate were associated with impaired signal transducer and activator of transcription (STAT3) and the nuclear factor kappa B (NF-κB) signaling pathway of loaded DCs. 19 By contrast, RNA could enhance innate immune reaction through TLR3, TLR7, and TLR8 signaling, using TRIF and MyD88 adapters. 39 DCs could uptake RNA via macropinocytosis, 40 and it could be translated into an endogenous antigen, which is further presented by major histocompatibility complex (MHC) class I directly to CD8+ T-cells, while exogenous protein antigen is presented by MHC class II to CD4+ T-cells. 41 These could explain our finding that RNA-pulsed DCs activated effector cells more efficiently than that of protein lysate–pulsed DCs against CCA cells.
To further investigate the effect of antigen pools on anti-CCA enhancement, we then combined mRNAs extracted from three distinct established CCA cell lines, including KKU-100, KKU-055, and KKU-213, to generate tumor antigen pools for DCs transfection. The effect of increased antigen diversity on DC maturation was determined (Figure 4). Similar to the previous experiment between protein lysate and RNA, DCs pulsed with either an individual or combined mRNAs were similar in expression of DC maturation markers (CD83 and HLA-DR) and co-stimulation molecules (CD86 and CD40). This indicated that loading of DCs with combinations of mRNA from up to three distinct cell lines did not significantly alter DCs maturation or co-stimulatory molecules.
Loading DCs with pooled mRNA was hypothesized to be beneficial to broaden and strengthen the killing capacity of effector immune cells. After recognition of peptide/MHC complex on a tumor, effector T-cells consequently lyse tumor cells by apoptosis induction. 41 Effector cell induction of CCA apoptosis was then determined (Figure 5). To better evaluate increased cytolytic activity, we reduced the E:T ratios from 5:1, 10:1, and 20:1 to 2.5:1, 5:1, and 10:1, respectively. Unlike the case with the KKU-100 cell line, the study of the KKU-213 cell line showed that DC pulsing with mRNA from the same target cell line failed to stimulate an adequate anti-tumor immune response. Based on our observations, we found that transforming growth factor beta (TGF-β) is highly expressed at the mRNA level in the KKU-213 cell line (unpublished data). This could limit DC function in antigen presentation in autocrine or paracrine fashion.42,43
For the broad-spectrum antigen, it has been investigated that pooled mRNA pulsing of DCs from multiple prostate cancer cell lines stimulated an anti-tumor immunity in prostate cancer patients. 21 However, the difference in efficacy between the one and multiple cell lines was not investigated. Our study was able to show that when mRNA from the KKU-213 cell line was combined with one or two distinct sources of mRNA for DC pulsing, they could induce a strong effector function against CCA. Significantly, a twofold killing enhancement of effector cells in DC loading with pooled mRNAs from three CCA cell lines was observed, as compared to mRNA from KKU-213 alone. Notably, cytolytic activity against cholangiocyte was not significantly changed, indicating the specific cytolytic activity against CCA cells.
Regarding mRNA combinations, amounts of 3 µg in this study provided better results, which successively decreased by reducing the combination amounts to 2 µg and 1 µg (Figure 5(b)). The study of Osada et al., 44 showed that an increased mRNA amount for DC transfection did not enhance anti-tumor cytotoxic T-cells in colon cancer. Moreover, Avci-Adali et al., evaluated green fluorescent protein (GFP) expression of cells transfected with different amounts of mRNA encoding enhanced GFP (eGFP). The result demonstrated that increasing mRNA amounts did not significantly increase transfection efficiency. 45 In our study, at equal amounts of mRNA (2 µg) but different mRNA combinations, the mixture of KKU-213 and KKU-055 for DC transfection was found to activate anti-tumor effector function more effectively than the mixture of KKU-213 and KKU-100. This suggests that the specific combination of mRNA from different cell line sources, not only the amount, is a factor influencing the anti-CCA immune response. Although the in vitro cytotoxic assay in this study has provided convincing results of CCA killing, it is still inadequate to ensure the anti-cancer immunity. Further in vitro and in vivo studies are required to evaluate the efficacy of RNA-pulsed DCs for CCA immunotherapy.
The carcinogenesis of OV-related CCA occurs by host chronic inflammation that promotes damage to DNA repair genes/proteins, proto-oncogenes, and tumor suppressor genes. 46 Thus, cytolytic activity that was observed in CCA cell lines was not attributable to the activity against antigens from OV. The study from our group showed that the expression level of at least five tumor-associated antigens (TAAs), including mucin 1 (MUC1), CD133, annexin A2, trefoil factor 1 (TFF1), and protein kinase A regulatory subunit 1 alpha (PRKAR1A) were different for their mRNA and protein expression among three OV-associated CCA cell lines used in this study (unpublished data). This might lead to different immunogenicity of a particular cell line for induction of the immune system.
Footnotes
Acknowledgements
Mutita Junking and Janya Grainok contributed equally to this work.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Ethical approval
The use of peripheral blood mononuclear cells (PBMCs) from healthy donors was ethically approved by the Siriraj Institutional Review Board, Faculty of Medicine Siriraj Hospital, Mahidol University (
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was supported by grants from The Foundation for Cancer Care Siriraj Hospital, the International Research Network (IRN) under Thailand Research Fund (TRF; No. IRN58W0001), Mahidol University, and TRF (No. IRG5980006); Charoemprakiat Funds and research grant (No. R015831045) from Faculty of Medicine Siriraj Hospital, Mahidol University; the TRF Grant for New Researcher (No. TRG5780173); the Newton Fund, Institutional Link; and the TRF-Royal Golden Jubilee (RGJ)-Ph.D. Scholarship (No. PHD/0044/2556).