
Abstract
From the application of Coley’s toxin in the early 1900s to the present clinical trials using immune checkpoint regulatory inhibitors, the history of cancer immunotherapy has consisted of extremely high levels of enthusiasm after anecdotal case reports of enormous success, followed by decreasing levels of enthusiasm as the results of controlled clinical trials are available. In this review, this pattern will be documented for the various immunotherapeutic approaches over the years. The sole exception being vaccination against cancer causing viruses, which have already prevented thousands of cancers. We can only hope that the present high level of enthusiasm for the use of immune stimulation by removal of blocks to cancer immunity will be more productive than the incremental improvements using previous immunotherapies.
Keywords
Introduction
The idea that the immune response could not only protect and defend us against infectious diseases but also provide a defense against cancer was developed in the late 1800s with concepts such as the side chain theory and targeted therapy (magic bullet) by Paul Ehrlich. 1 The approached championed by Ehrlich was applied to cancer chemotherapy as well as to cancer immunology. 2 The direction of the immune response to infectious diseases using vaccines (vaccination) has contributed immeasurably to the health and longevity of humans because of protection not only against viral infections 3 but also against bacteria, 4 fungi, 5 and worms. 6 As presented in the following, vaccines against viral infections that cause cancer such as hepatitis B and human papillomaviruses are very effective in preventing cancers of the liver and cervix (see below). In this review, many of the attempts to use the immune system against cancer are presented. These varied and diverse approaches first produced striking anecdotal results with reports of individual patients with complete cures or substantial remissions. This pattern has been repeated many times with the results of disappointing but incremental advances in the treatment of cancer. There are high expectations that removing blocks to immunity (checkpoint inhibitors) will allow the immune response to individual cancers to attack and either cure or reduce the growth of the cancer. The question remains will this be the long anticipated breakthrough or will we again see an incremental advance in cancer therapy.
Coley’s toxin
The saga of cancer immunotherapy begins in the 1890s with the development of Coley’s toxin by Dr William B. Coley described in detail by Stephen Hall in “A commotion in the blood” 7 (see also review by McCarthy 8 ). Shortly after receiving his license to practice medicine in New York in 1890, Coley observed that occasional patients with inoperable cancers had remissions of their tumors during life-threatening infections such as severe erysipelas (Streptococcus pyogenes). In 1891, he intentionally infected a patient with S. pyogenes, who had a large inoperable neck tumor. After a life-threatening systemic infection, Mr Zola recovered and his tumor showed marked regression. By 1891 he had treated three patients with large tumors; all had noticeable reduction in tumor size, but two of the three patients died. 9 During the next few years, Coley intentionally infected 12 additional cancer patients: 4 developed severe infections, 2 died, and 2 appeared to be cured of their tumors. Eight other patients did not develop severe infections and tumor reduction was seen in none of these. 10 In 1893, Coley produced tissue extracts of cultures of S. pyogenes and Bacillus prodigiosus (an endotoxin producing organism) that became known as Coley’s toxin. Although not known at the time, this toxin acts as a superantigen and activates release of cytokines, including tumor necrosis factor (TNF), interferons (IFN), and a number of interleukins (ILs). Treatment of a 13-year-old boy with inoperable metastatic tumors by direct injection of the toxin into the tumors resulted in severe fever and chills and the tumors shrank. This patient lived to the age of 47 years and died of causes other than cancer. In 1910 in a lecture at the Royal Society of Medicine in London, Coley showed photographs of a patient with a large tumor on the face that became much smaller after 63 injections of his toxin. 11 By 1914 Coley 12 had treated over 500 patients, claiming to have induced over 150 remissions. However, other physicians could not reproduce his results. Because of this failure, the potentially fatal side effects of toxin administration, and the introduction of irradiation for cancer treatment, the toxin approach was discontinued. Coley’s positive experience with early positive results within a limited number of patients, followed by an inexplicable inability to duplicate the results, establishes a pattern that besets those working in cancer immunotherapy to this day (Figure 1). It is likely that Coley induced what is today known as a cytokine storm. 11 The severe fever and chills are reproduced today after treatment in some patients, and the specific cytokines that appear to be involved include IL-2, TNF-β, IL-12, IFN-α, and TNF-α. Each of these was used in clinical trials in the 20th century as is reviewed below. The next approach extends the use of bacterial stimulation to activate the cell-mediated arm of the immune response to become active against cancer.

Projected enthusiasm levels for various cancer immunotherapies. As early anecdotal reports of spectacular cures of patient with advanced cancer appear (usually in the press), there is high enthusiasm, remarkable press coverage, and large amounts of money raised for clinical trials. With phase I–II clinical trials, the response rate substantially decreases and enthusiasm begins to decline, but press reports continue to be high. After phase III trials, a relatively modest response for limited type of cancer is reported (see Table 1). Finally, because of dangerous side effects and limited long-term remissions, general clinical application is limited.
Bacille de Calmette de Guerin
Bacille de Calmette de Guerin (BCG) is an attenuated form of tubercle bacillus used with limited success as a vaccine for tuberculosis. A large-scale epidemiologic study in Europe suggested a slight decrease in the incidence of cancer in population immunized with BCG, compared with populations not immunized. This application of BCG is not recommended in the United States at this time. In the early 1960s, Bast et al. 13 at National Institutes of Health (NIH) demonstrated complete remission of transplantable hepatomas in guinea pigs treated with BCG. Injection of the tumor cells into the flank led not only to local growth of the transplanted tumor but also metastasis to the draining lymph node. Injection of BCG into the primary transplant site resulted in infiltration of lymphocytes around the tumor sites with regression of both the primary and metastatic lesions. It was then reported that this approach could be used to induce regression of leukemia and melanoma lesions in children, but such results were again not reproducible. On the one hand, repeated injections of BCG into the bladder of humans with superficial bladder cancer have led to significant remissions. 14 This application via the bladder produces recruitment of activated lymphocytes and macrophages into the bladder wall, increases ILs in the urine, and induces systemic immunity to BCG. Long-term remission is achieved in up to 50% of patients; about 35% show no response and 5% have severe adverse effects. 15 Overall, as with Coley’s toxin, the pattern of response of cancer to BCG did not meet the enthusiasm of the original reports but at least an incremental advance was made by this approach in the treatment of a specific type of tumor, superficial bladder cancer. The non-specific activation of cell-mediated immunity was then attempted using contact sensitivity haptens that would activate T-cell cytotoxicity to superficial cancer cells exposed to the hapten.
Dinitrochlorobenzene
Dinitrochlorobenzene (DNCB) acts as a hapten that binds to tumor cells in the skin and induces a T-cell cytotoxic response analogous to poison ivy (contact dermatitis). Painting superficial skin metastases with DNCB induced a marked local inflammatory reaction at the lesion sites. 16 At first, it was reported that treatment of the skin lesions was followed by reduction in the size of internal metastases. However, this was not reproducible. While DNCB treatment appears to have some cosmetic effect, there is little to no evidence to indicate that there is an effect of overall survival. Treatment of therapy-resistant warts with DNCB appears to have a beneficial effect. 17 If non-specific cell-mediated immune did not work, perhaps the effector cytokines could be used.
Non-immune-specific cytokines
IFN-α
Viral interference, discovered in the 1930s, results in protection of cells or animals against a viral infection, if there has been a recent previous infection with a less severe, unrelated virus. Alex Isaacs and Jean Lindenmann 18 found that the interference phenomenon could be transferred from treated cells to uninfected cells by tissue culture fluid. They called the active factor IFN and later showed that it was produced by the cultured cells and not by the virus. Later, this was known as IFN-α when Gresser et al. 19 in 1969 found a new IFN made by lymphocytes, IFN-γ. In 1966, Gressner found that IFN-γ not only inhibited virus-induced tumors but also reduced growth of epithelial tumors. Initial reports of substantial remissions or different cancers were followed by more complete studies that showed little or no effect. In 1986, Jorge Quesada et al. 20 at the MD Anderson Cancer Center treated a 25-year-old man with advanced hairy cell leukemia with IFN-α. At that time, there was no effective treatment for this disease. Daily doses of 3 million units achieved complete remissions. By 1986, IFN-α had been used to treat 30 other patients with this disease, achieving 9 complete and 17 partial remissions. IFN-α was subsequently approved for treatment of hairy cell leukemia by the U.S. Food and Drug Administration (FDA).21,22 Although not effective for other cancers, the action of this agent reinvigorated the field of cytokine treatment of cancer.
TNF
In 1975, Carswell et al. 23 found that a novel factor appeared in the serum of mice after treating with BCG and endotoxin had induced necrosis of tumors; they called this substance TNF. Another effect of this factor, independently discovered by others, was to cause wasting disease in treated animals with large tumors. Because of this effect, TNF was also called cachectin. In other trials, TNF produced sever systemic effects (cytokine storm) including fever, chills, and systemic shock. At the maximum tolerated dose, there was little, if any, effect on tumor growth. Thus, the prospects for using this very interesting and potent factor for treating cancer were not promising.
In 2006, Ferdy Lejeune et al. 24 infused 10× the maximum tolerated dose of TNF into the isolated limb of a patient with multiple melanoma lesions and the lesions faded away. The systemic shock that was caused by TNF that escaped into the systemic circulation had to be aggressively treated. Later, the treatment effect was found to be enhanced by adding low-dose IFN-γ and chemotherapy (Mephalan). 25 This was known as “triple therapy.” This approach has limited applications because it can only be used on isolated limbs 26 . Lejeune stated that “You can cure the limb, but you can’t cure the patient. The current use of TNF in cancer is in the regional treatment of locally advanced soft tissue sarcomas and metastatic melanomas or other non-operable tumors in order to avoid amputation of the limb. 27 These results led to further cytokine/cell-mediated approaches.
IL-2 and natural killer cells
In the 1960s, it was observed that supernatants from cultures of lymphocytes stimulated with mitogens, such as phytohemagglutinin (PHA), produced factors that had biological effects on other cells (conditioned medium). These factors were given names that reflected their biological effects, such as migration inhibitory factor (MIF) for macrophages, macrophage activating factor (MAF), lymphocyte activating factor (LAF), and lymphocyte migration inhibitory factor (LIF). In 1974, supernatants of PHA were used as conditioned medium to stimulate growth of cultured leukemic cells. The stimulatory factor in the conditioned medium was first named LAF and then as T-cell growth factor and IL-2. In 1986, Steve Rosenberg and Lotze 28 at NIH began to organize a team to develop immunotherapy of cancer. He started with passive transfer of spleen lymphocytes from pigs inoculated with tumor cells from selected patients (refer adoptive transfer explained in the following). As no positive results were found, he looked for a new approach. Also at NIH, Elizabeth Grimm 29 attempted to culture T-cells with cancer cells and IL-2 in vitro to obtain a tumor-reactive T-cell line. She did obtain lymphocytes that could kill tumor cells, but these did not have the properties of T-cells. These were called lymphokine activated killer cells (LAKs). Many years earlier, Karl-Eric and Ingegard Hellstrom et al., 30 in Seattle, had cultured pediatric neuroblastoma cells with lymphocytes from the same patients to identify immune cells from the patients that would inhibit growth of the tumor cells (colony inhibition assay). Their preliminary findings suggested that some patients with remission had lymphocytes that inhibited colony formation. However, this inhibition was significant only when compared to a group of control lymphocyte donors whose lymphocytes had no effect on colony formation. When other normal donors were used as a cell source, their lymphocytes did inhibit tumor growth even though there was no evidence that they had been previously exposed to the tumors. This effect was proposed to be due to non-immunized normal lymphocytes that were named natural killer cells (NKs). 31 Grimm 29 found that IL-2 exposure increased the activity of NKs, designating these cells as LAKs.
In the meantime, Rosenberg and Michael Lotze 28 attempted to use large amounts of Il-2 for immunotherapy. By 1984, 75 patients had been treated with IL-2 plus LAKs with little or no response. If the dose of IL-2 was increased to the maximum tolerated by the patients, several spectacular cures were found. However, these patients developed severe side effects, including vascular leak syndrome and life-threatening pulmonary edema. These effects were so severe that treatment had to be stopped. Two patients given up for dead actually recovered and were judged to be free of cancer. Biopsies from tissues of successfully treated patients showed lymphocytic infiltration and necrosis, and over the next 3 months, the tumor disappeared. In studies of an additional 45 patients, a response rate of up to 45% was reported with LAK/IL-2 treatment. Again it appeared that a breakthrough in cancer therapy had been made. Observations by Rosenberg and co-workers received national lay press coverage. When more complete studies were available, complete responses to this regimen in 8% of patients and partial responses in another 10% of patients were found. However, severe side effects including shock (capillary leak syndrome), chills, fever, nausea, vomiting, diarrhea, cutaneous erythema, anemia, and moderate-to-severe liver and kidney dysfunction as well as thyroiditis cause most centers to discontinue this approach.32,33 As of today, IL-2 is only approved for treatment of mycosis fungoides and Sézary syndrome, tumors of mature T-helper cells with less than satisfactory results. A recent study of these conditions recommends IFNs, retinoids, and chemotherapy (doxorubicin and gemcitabine) as well as a monoclonal antibody, alemtuzumab (anti-CD52), but not IL-2. 34 A more recent approach is the use of IL-2 fused with the diphtheria toxin, a chain which delivers the toxin to the IL-2 receptor on the T-helper cell (CD25). 35 IL-2 is still used in trials in combination with other therapies such as adoptive transfer of lymphocytes to stimulate T-cell proliferation (see in the following) in otherwise untreatable cases. Overall, these results are very similar to those previously reported for Coley’s toxin, and in fact, it is likely that Coley’s toxin did produce high levels of IL-2. Now, treatment approaches were then extended to the use of other ILs.
IL-12
IL-12 is produced by activated antigen-presenting cells and has pleiotropic effects. 36 Originally called natural killer cell stimulatory factor (NKSF), 37 IL-12 not only activates NK cells but also has other actions. These include activation of CD4+ Th1 helper cells and CD8+ T-cytotoxic cells, stimulation of B-cells, inhibition of angiogenesis, and blockade of growth of human tumors in immune-deficient mice, particularly if the IL-12 gene is introduced into fibroblasts. 38 In early FDA phase I clinical trials (testing for toxicity and dose level that can be given), no problems were found when patients were primed with low doses of IL-12 and then given daily doses of IL-12 2 weeks later. However, in phase II trials in 1995, higher doses of IL-2 were given. Of 15 patients treated with high doses of IL-2, all became seriously ill (cytokine storm) and 2 died. Because of high toxicity, IL-2 is now used in clinical trials as an addition to gene therapy and only as local injections.39,40 The robust response seen in animal studies has not been translated for humans. If one cytokine gave less than satisfactory results, perhaps more than one cytokine would be more effective.
Cytokine mixtures
Doses of toxic cytokines, such as IL-2 and TNF, may be given in mixtures at lower than toxic levels. An example of such a mixture is a commercially available preparation of natural cytokines called “leukocyte IL” or “Multikine” (Cel-Sci Corporation, Vienna, VA, USA) that includes IL-2, granulocyte-macrophage colony-stimulating factor (GM-CSF), IFN-γ, and TNF-α. 41 Phase II trials suggest that Multikine may stimulate a healthy immune system. However, a phase III clinical study in head and neck cancer has been put on hold by the FDA. While cytokines were being extensively used, there was an increasing interest and application of transfer of immune active cells.
Adoptive cell transfer
Tumor-infiltrating lymphocytes
In 1894, the preeminent American surgeon at Johns Hopkins, William Halsted, 42 noted that the breast cancer patients whose tumors had a lymphocytic infiltrate had a better post-operative prognosis than those patients whose tumor did not have an infiltrate. He postulated that these lymphocytes were reacting against the tumor and were inhibiting growth. However, an alternative hypothesis was presented that the reason that the tumors with infiltrates did not metastasize as readily as those without was because such tumors required stimulation by the lymphocytes, and thus, they were more dependent on the lymphocytes for their growth than those without lymphocytes (immune stimulation).43,44 The effect of lymphocytes on tumors may depend on the type of infiltrating lymphocytes. For example, infiltrates of Th1 cells indicate a good prognosis, whereas infiltrate with Th2 suggests a poor one. 45 In any case, such lymphocytes became known as tumor-infiltrating lymphocytes (TILs). In 1984, Alexander Knuth et al. 46 extracted lymphocytes infiltrating a tumor, cultured them with IL-2, and showed that they could lyse tumor cells. These cells were T-cytotoxic cells and were specific for the tumor from which they were isolated. Rosenberg at NIH soon reported that reinfusion of TILs back into the patient from induced “substantial remission” in eight or nine patients with melanoma. After treatment of more patients, a partial response rate of 55% was claimed,28,47 and in subsequent studies, a much lower response was found. This approach has been abandoned. A next step was to transfect the TILs with cytokine genes that might increase the efficiency of TILs. This has not proven effective. A more recent approach being tested is to insert chimeric tumor antigen receptors on peripheral blood lymphocytes cultured from the patient. 48
Chimeric antigen receptor T-cells
In this approach, T-cells from the blood of patients with refractory B-cell tumors are transfected with anti-CD19, so that they recognize essentially all stages of B-cell development.49,50 Upon transfusion into the patient, these cells can effectively attack B-cells. The more recent versions of these engineered cells contain extracellular domains that recognize CD19 linked to intracellular activation domains which when the receptor binds to CD19 stimulates proliferation and cytokine production by the transplanted cells. They have been used effectively for management of acute lymphoblastic leukemia and are proposed for use in Hodgkin’s disease and blast crisis of chronic lymphocytic leukemia. The transfused cells rapidly expand in the patient. 51 This is desirable because the result is that there are more effector cells to attack the cancer. This is undesirable because these cells also attack normal B-cells, causing severe clinical complications including B-cell aplasia, neurologic toxicity, cytokine release syndrome, allergy, and anaphylaxis. This has led to consideration of how to diminish or prevent the side effects. 52 An ingenious approach is to construct the chimeric receptor T-cell so that the activation domains of the engineered receptor require a small molecule to join them.53,54 Thus, control of the CAR T-cells can be manipulated, so that the activity can be turned on when necessary. It remains to be seen how much of an impact this approach will eventually have. First, it is presently applied to a small proportion of leukemia patients because about 80% of patients with B-cell cancers respond well with prolonged complete remissions to standard chemotherapy and/or monoclonal antibody treatment without CAR T-cells (see in the following). Second, the serious side effects may eventually prove to be worse than the treatment for many patients. Third, obtaining peripheral blood lymphocytes from each patient, rapidly transferring them to production laboratories for culture, and genetically engineering the cells are prohibitively expensive, unless some advance in methodology occurs. Thus, this approach is an example of impressive biomedical engineering, but it may simply be impractical for more than occasional use (Figure 1). For more information, see the article on the front page of the New York Times (2 August 2016).
Immune RNA
The rationale for this approach was that the RNA could be extracted from immune cells of immunized animals (sheep) and used to transfer immunity to cancers to which the sheep was immunized. 55 There are problems with this approach. First, if the immune RNA is active, then immunity to the normal cells of the patients would most likely be transferred as well. However, if specific tumor immunity could be induced, the injected RNA would be rapidly destroyed by the potent ribonucleases present in blood and tissue fluids. 56 An interesting sidelight of this work is that chest X-rays of lung cancers before and after treatment were used to evaluate the size of the tumor seen. However, not only the size of the tumor was smaller but also the size of the heart and the density of the ribs were lower. 55 Thus, it has been suggested that the size of the tumor on the chest X-ray was determined by the extent of radiological exposure. Better success will result through the use of active stimulation of the immune response to tumor-causing viruses.
Tumor vaccines
Viral vaccines
With considerably less fanfare than other immunological approaches, vaccination against cancers caused by viruses has been remarkably successful. 57 Thousands, if not millions, of serious cancers have been prevented by this approach. This list of viruses and virus-induced cancers includes Epstein–Barr virus (Burkett’s lymphoma, etc), hepatitis B virus (HBV, hepatocellular carcinoma (HCC)), hepatitis C virus (HCV, hepatocellular carcinoma, human papillomavirus (cervical cancer, etc.)), Kaposi’s sarcoma virus, and Merkel cell carcinoma virus. 57 Worldwide, over 2 billion people are infected with HBV. HBV infection is believed to be responsible for up to 60% of HCC. 58 In endemic areas of HBV infection, introduction of HBV vaccines has reduced the prevalence of chronic infection in children by more than 90%. 59 Thus, vaccination against HBV has already had a significant effect in reduction of HCC and cirrhosis. There is no doubt that vaccination against viruses causing cancers has had more impact on preventing cancers than any other immunological approach. If vaccination against tumor-causing viruses works, then maybe vaccination against tumor antigens would be effective.
Vaccines against tumor-specific antigens
Self-healing malignant melanoma
The occasional observation of spontaneous remissions of cancer gives rise to speculations of the development of a cancer-specific immune response in such patients. Figure 2 shows the expanding growth of a self-healing malignant melanoma, while that the central zone of the tumor shows healing with no melanoma lesion. Histologically, lymphocyte infiltration of the tumor may be observed along the regressing margins, with collections of melanin-containing macrophages underlying the normal appearing skin in the center of the lesions. Since 1866, there have been 76 reported cases of spontaneous regression of metastases of malignant melanomas. 60 The mechanism of these regressions could be immunological, endocrine related, or related to loss of tumor vasculature. 61 However, the histological findings are consistent with a delayed-type cellular response to the tumor and suggests tumor immunity. Most studies of tumor-specific immunity in patients address malignant melanoma or renal cell carcinoma, two tumors noted for their relatively high incidence of spontaneous remission of metastases as compared to other cancers. Direct evidence for tumor-specific transplantation antigens was originally derived from tumor transplantation studies in mice.
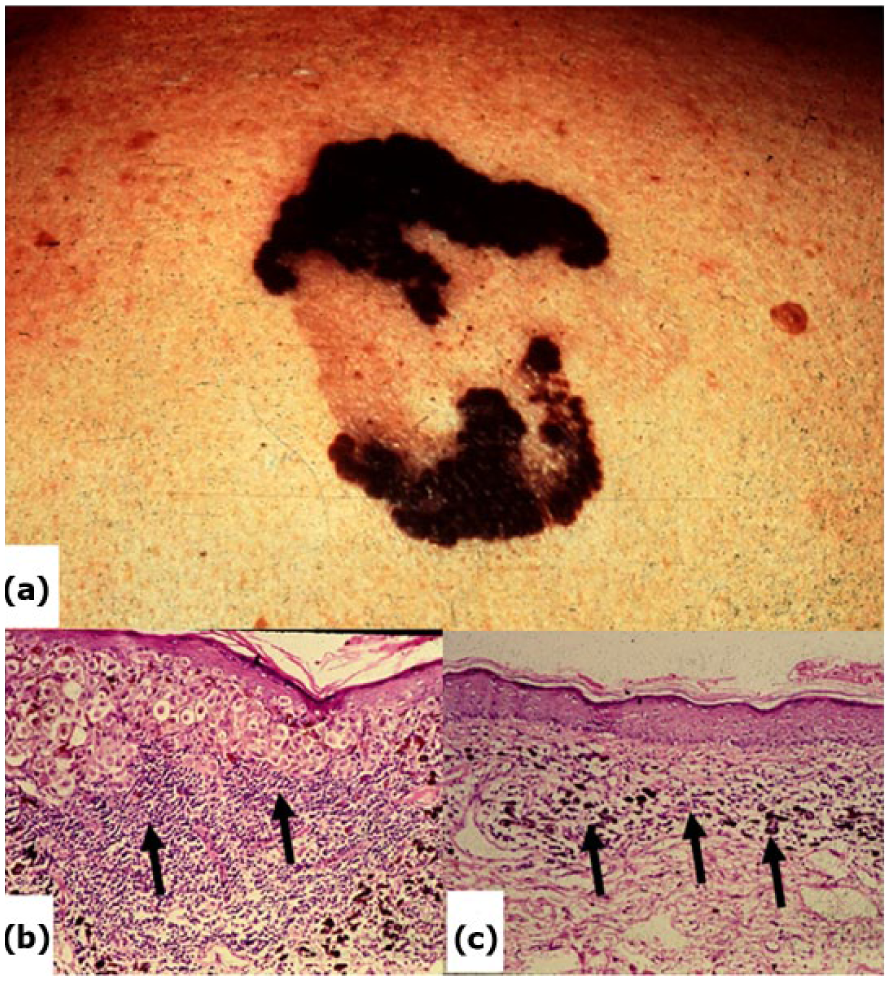
(a) Gross photograph of self-healing malignant melanoma. The growing margins of the cancer are advancing at the same time that the center has “healed.” (b) Lymphocyte infiltrate underlying the cancer. (c) Histocytes (macrophages) containing melanin underlying the “healed” zone.
Tumor-specific transplantation antigens
In the 1890s, Paul Ehrlich 62 studied transplantation of tumor cells in animals and noted that growth of metastatic lesions increased after removal of the primary tumor. He attempted to generate immunity to cancer by injection of “weakened” cancer cells. During the early part of the 20th century, it was found that tumors transplanted from one mouse to another were rejected. This, of course, is because of histocompatibility differences (graft rejection). In 1943, Ludwig Gross 63 observed that tumors were rejected more slowly in partially inbred mice, and a decade later, Foley64,65 showed that tumor grafts growing in inbred mice were rejected by mice immunized to the tumors. In 1960, George Klein et al.66,67 did the definitive study to show that tumors did have tumor-specific transplantation antigens (Figure 3). 68 Several fundamental concepts were observed from studies of specific tumor transplantation.

Demonstration of tumor-specific transplantation antigens by Geroge Klein et al. (a) Primary methylcholanthrene–induced cancers from inbred mice were excised and the cells maintained in vitro. (b) When these cells were injected into another mouse of the same strain, they grew to produce tumors. However, when injected back into mouse A from which the primary tumor had been removed they did not grow. (c) Then, when the primary mouse was injected with cells from a tumor arising in a different mouse of the same strain, these cells grew. The conclusion is that the original mouse developed transplantation immunity to the primary tumor that is specific for that tumor and not for other tumors of the same type: tumor-specific transplantation immunity.
Concomitant immunity
A tumor bearing individual may be immune to his/her own tumor cells. Tumor cells taken from an individual’s own tumor will not grow if injected into another site even though the tumor continues to grow at the primary site. 69
Immunosurveillance
Newly arising tumors are recognized by the immune system and held in check by immune mechanisms.70,71 Of interest, however, there is no increase in tumor incidence in immunodeficient mice, which cannot mount an immune response to cancer. 72
Immunoediting (also known as immunoselection)
The immune systems select for destruction immunogenic tumor cells. The result is that the tumors that do arise are weakly antigenic or non-antigenic. 73
Human tumor-specific vaccines
The first human tumor-specific antigen vaccine was developed after the identification of a tumor antigen on malignant melanoma cells by Theiry Boon in 1993. 74 In 1981, Alexander Knuth in Germany sent cancer cells from a patient with multiple metastases to Boon in Belgium. Boon cultured the cells in vitro and treated them with mutagens. After irradiation, the non-viable melanoma cells were injected back into the patient. After three injections of such cells over 3 months, the patient’s tumors disappeared and with repeated injections did not recur. Lymphocytes harvested from the patient very effectively killed the cultured tumor cells in vitro. Boon then used IL-2 to stimulate growth of a T-cell line from the patient. When the T-cell line was tested against a DNA library from tumor cells transfected into tissue-cultured cells, the cloned T-cell line killed some of the transfected cells. From one colony of susceptible cells, a 9-amino acid peptide was identified, which was presented as an antigen by class II major histocompatibility complex (MHC). This peptide was designated as melanoma-associated antigen-1 (MAGE-1). Since then, a number of putative tumor antigens have been identified from melanoma, prostate, pancreas, and colon tumors. 75 Unfortunately, most of these have had very little effect on tumor growth, but some positive effects have been obtained in combination with chemotherapy. 76 For example, vaccines for prostate cancer increase survival in metastatic castration-resistant cancer (Sipuleucel 4.1 months; Prostvac 8.5 months). 75 Glycoprotein 100 (gp-100) or melanocyte protein PMEL is a 661 amino acid transmembrane glycoprotein that is enriched in melanosomes. When applied topically, it permeates mouse and human skin and induces T-cell cytotoxic responses. 76 It is currently being tested as an adjunct to chemotherapy in some clinical trials. It appears that high levels of antigen-specific T-cells do not necessarily prevent progression of human melanomas. 77 Thus, although there may be some beneficial effects in combination therapies, tumor vaccines do not play a prominent role in immunotherapy. Paul Ehrlich’s 1 original hypothesis on the side chain theory predicted antibodies as the magic bullet for immunotherapy. This theory is being tested using monoclonal antibodies.
Monoclonal antibodies
In 1975, Georges Kohler and Cesar Milstein 78 fused antibody-producing B-cells with cells of multiple myeloma to form an immortalized cell line that produced high levels of the specific antibody. This Nobel Prize winning discovery allowed for the production of large amounts of a single antibody (monoclonal antibody, -mab). In 1984, Masui et al. 79 at University of California, San Diego (UCSD) demonstrated that administration of a mouse monoclonal antibody to the human epidermal growth factor receptor (EGFR) could markedly inhibit the growth of human cancer cells in immunodeficient mice. The mouse model proved the principle, but mouse monoclonal antibodies cannot be used in humans due to their high immunogenicity. Subsequently, a massive and incredibly productive effort was made to genetically engineer antibody-producing cells using recombinant DNA to create constructs capable of expression in mammalian cell lines. 80 Gene segments capable of producing antibodies were isolated and cloned into tissue culture cells that could be grown in a bioreactor. The antibody proteins produced from the DNA clones were then harvested in large amounts. This was a major advance, but even more astounding was that the DNA could be engineered to express different parts of the antibody molecule of mice and humans. Thus, the genetically engineered antibodies may have human constant chains and mouse variable chains; it may have human heavy and light chains, but the complementary determining regions (antigen binding sites) of the mouse antibody. In addition, fully human antibodies have been produced (see Figure 4). These products have been coded by specific suffixes. That is, -mab refers to monoclonal antibody, -omab refers to mouse monoclonal antibody, and -zumab refers to humanized mouse monoclonal antibodies.
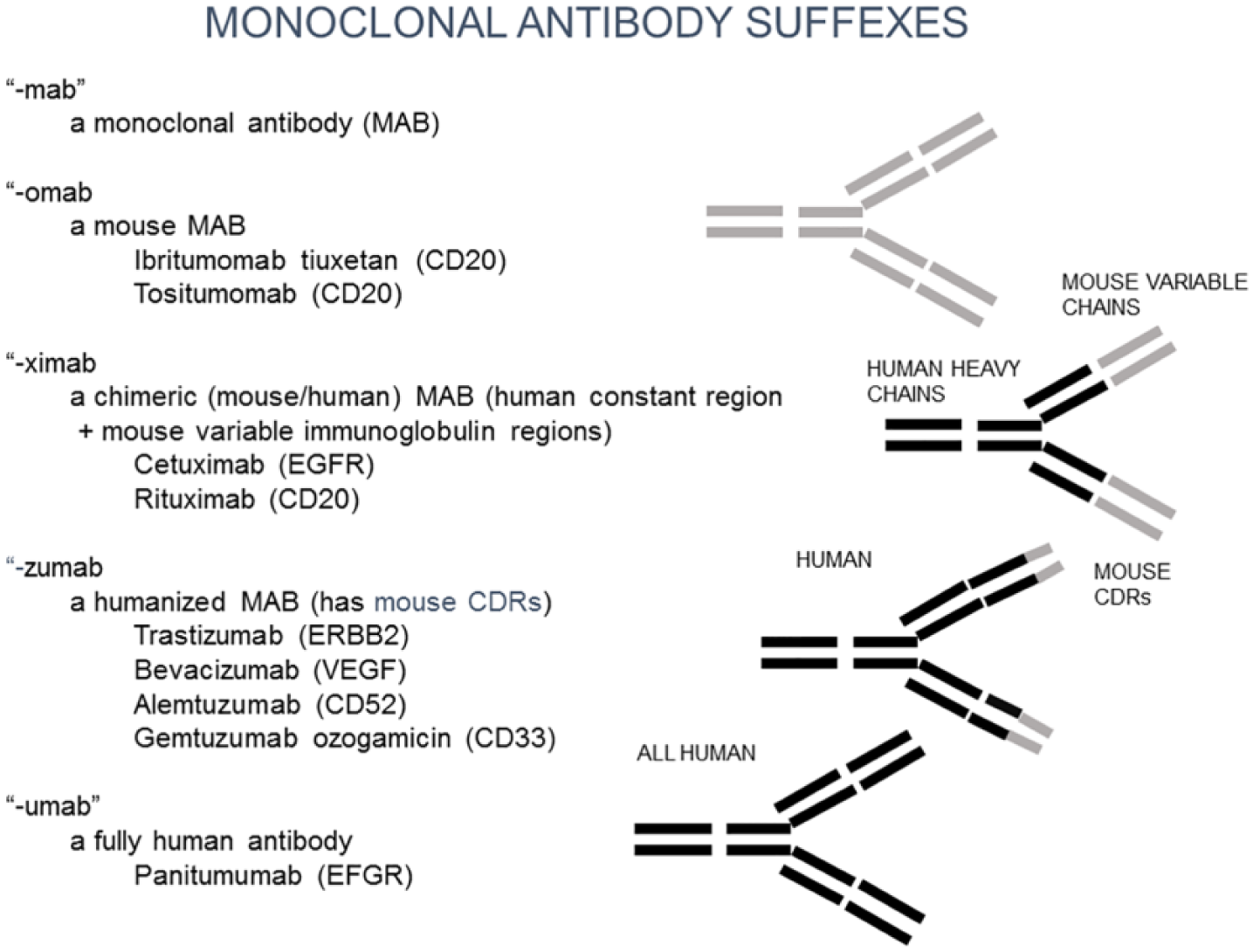
Genetically engineered monoclonal antibodies. Because mouse monoclonal antibodies cannot be used for human therapy, various parts of the antibody are replaced by human immunoglobulin chains. These are identified by suffixes: -omab is a mouse monoclonal; -ximab has human constant heavy and light chains and mouse variable heavy and light chains; zumab (humanized) has human chains except for mouse complementary binding regions (antibody binding sites); -umab is a fully human antibody.
Rituximab
One of the first anticancer antibodies to be used was the human B-cell marker CD20 (rituximab) for treatment of B-cell malignancies. Rituximab has significantly improved the treatment of diffuse large-cell B lymphoma and follicular lymphoma. 81 When combined with fludarabine and cyclophosphamide, rituximab treatment has achieved long-term disease-free survival in chronic lymphocytic leukemia associated with mutated immunoglobulin heavy chain variable region. 82 About 70% of patients with acute B-cell tumors have complete remissions with the combination of cyclophosphamide, doxorubicin, vincristine, and prednisone therapy (CHOP) plus rituximab, whereas the remission rate is 40% when CHOP is used without Rituximab. 83 As is the case in other immunotherapy approaches, success of this regimen is limited to selected cancers.
Herceptin (trastuzumab)
Herceptin is a humanized mouse monoclonal antibody to the human epidermal growth factor receptor 2 (HER2) present on about 30% of human breast cancers and certain colon cancers. 84 This antibody is used in conjunction with chemotherapeutic regimen of anthracycline, cyclophosphamide, or paclitaxel. Herceptin blocks the cell signaling necessary for continued growth of the cancer cells. Adding herceptin to chemotherapy increases disease-free survival about 10% over chemotherapy alone (84% vs 75%). This is achieved at a cost of increased side effects, such as congestive heart failure and acute leukemia. 85
Other monoclonal antibodies
Overall, over 300 genetically engineered monoclonal antibodies have been approved by the FDA for human use in a variety of diseases in addition to cancer. 86 Among the other cancer-directed monoclonal antibodies are alerntuzumab (targeted at CD53) for chronic lymphocytic leukemia, gemtuzumab (directed at CD33) for acute myelogenous leukemia, and cetuximab, panitumuab (EGFR), and bevacizumab (vascular endothelial growth factor (VEGF)) for colon and lung cancer. 86 In general, FDA phase II trials showed promise for anti-EGFR therapies, which was not the case in phase III trials. Trials continue to test this anticancer approach in combination with chemotherapy. Results of monoclonal antibody treatment of cancer fit the pattern of high initial enthusiasm and eventual limited effectiveness in clinical trials. The field of tumor immunity continues to move to new ideas. The next idea is that there is an immune response to cancer, but it is inhibited by immune checkpoint regulators. If these checkpoints can be removed or attenuated, then an individual’s own anticancer response may be able to block growth of the tumor.
Immune checkpoint regulation inhibitors
Cytotoxic T-lymphocyte-associated protein 4
Editorial comments in the scientific and lay press have recently emphasized breakthrough developments in the very expensive trials of immunotherapy. These articles referred initially to discoveries that immune checkpoint regulation inhibitors (CPRIs) could be manipulated by specific antibodies.87,88 The rationale for this approach is that cancers can turn off the immune effector T-cytotoxic cells. Specific T-cells are activated by tumor-specific antigenic peptides presented by dendritic cells to the T-cell receptor (TCR) and by co-stimulation by reaction of B7 on the dendritic cells with CD28 on the T-cell. 87 However, IFN-γ production by the T-cell upregulates production of cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) in the cancer cell causing displacement of CD28 on the T-cell. Reaction of the TCR with MHC and CTLA-4 instead of CD28 with B7 on the dendritic cell results in death of the reactive T-cell and protection of the cancer cell. Treatment with anti-CTLA-4 blocks this inhibition and allows the CD28 to reappear on the T-cell, so that it can develop an immune response to the cancer. An antibody that can accomplish this is ipilimumab. The preclinical model for this approach was developed by Leach et al., 89 who showed that treatment of mice injected with anti-CTLA-4 greatly inhibited the growth of transplanted tumor cells. Early trials using anti-CTLA-4 (ipilimumab) showed that after 3 months of treatment, there was increased survival of patients treated with ipilimumab compared to patients treated with tumor antigen gp 100. Anti-CTLA-4 treatment achieved a plateau of survival of about 20% versus none in patients treated with gp 100 alone. 90 It should be pointed out that these were advanced cases for which other therapies had failed or showed little effect. In one study, treatment with trastuzumab increased responses to CTLA-4 blockade leading to increased T-cell responses in mice with primary breast cancers; there was, as well with cytotoxic T-lymphocyte antigen-4 (CTLA-4) blockade, an increase in Tregs, which are essential to protect mice from immune-mediated autoimmune reactions during tumor rejection. This effect is not so obvious in human subjects who may have decreased Treg activity and lymphocytic infiltration of various organs and symptoms of cytokine storm. 91 Predilection to severe side effects was supported by findings in the mouse model that included depigmentation and deaths of older mice that were apparently due to autoimmune reactions. 92
Programmed cell death protein-1/programmed cell death ligand-1
A second costimulatory immune checkpoint inhibitor is programmed cell death ligand-1 (PD-L1). 93 When antigen-specific T-cells react with antigen on tumor cells via the TCR, tumor-derived PD-L1 protein reacts with programmed cell death protein-1 (PD-1) and eliminates T-cell killer effects. Like anti-CTLA-4, anti-PD-L1 blocks this tumor defensive reaction and allows T-cells to attack the cancer cells. Zou et al. 94 recently reviewed the results of 22 ongoing clinical trials using variations of this approach. The clinical response rate varied from 14.5% in refractory non-small-cell lung cancer to 87% in refractory Hodgkin’s lymphoma (Table 1). In a comparison with chemotherapy alone, anti-PD-1 (Keytruda) treatment resulted in an increase of 17% in the 1-year survival rate of non-small-cell lung cancer. 95 Active investigation is underway to predict which patients will respond. 96 There is good correlation between the immunohistological identification of high expression of PD-L1 on the cancer cells and clinical response to anti-PD-1. 97 Other possible response markers are high levels of Th1 cytokines, the presence of tumor antigens, 98 lack of immunosuppression, high levels of PD-L1-high cytotoxic T-cells in tumors, 99 low cancer stem cell–like properties, 82 and mismatch repair deficiency. 98 The largely anecdotal cases of complete lasting clinical responses have achieved notoriety (New York Times, 31 July, 1 August 2016). Is this a true breakthrough or another incremental improvement in cancer immunotherapy? It is far from clear. The treatment is extremely costly, up to US$250,000 per year per patient. This approach has works in only a minority of patients, and there are potentially severe side effects. 100 This side effect profile is the result of blocking of controls on the immunoreactive T-cells, so that the T-cells also attack normal organs. As a result, a significant number of patients successfully treated for cancer develop life-threatening diabetes, cytokine storm, and a variety of autoimmune diseases.100,101 A major effort is now underway to try to prevent or limit these untoward reactions. However, they do present serious problems for the general application of CPRI therapies particularly outside of major medical centers. It would clearly be desirable to target the PD-1/PD-L1 checkpoint exclusively for cancers or non-immunologically downregulated expression of the PD-L1 gene in tumor cells. 102
Some examples of response rates in ongoing clinical trials with anti-PD-1 Nivolumab (Optivo).

Source: Modified from Thompson et al. 82
These are ongoing trials; final conclusive results are not yet available. Frequently, an early promising complete response is followed by resistance to treatment.
Combinatorial therapies
In the last 5 years, the number of clinical trials including combinations of chemotherapy, monoclonal antibody, tumor antigen, and immunotherapy has increased dramatically and is projected to continue to rise even further. 102 In fact, many of the early clinical trials of CPRI therapy were done on patients whose cancers had resisted conventional chemotherapy. 94 The projected potential combinations will keep clinical trials ongoing for many years. This includes combining chemotherapy and CPRIs with tumor antigen (gp-100), adoptive cell transplantation, targeting cancer stem cells and tumor stroma, microbiota alteration (selecting beneficial bacteria), activation of antigen-presenting cells, and signal induction inhibitors, such as BRAF and MEK inhibitors.103,104
Oncolytic virus immunotherapy
In this new variation on an old theme of viral oncolysis, viruses are engineered to infect specific tumors and cause their destruction. 105 The FDA approved in 2015 an injectable form of T-VEC, Imlygic, for the treatment of melanoma in patients with inoperable tumors. 106 T-VEC is a re-engineered herpes simplex virus 1 (HSV-1), a relatively innocuous virus that normally causes cold sores. Genetic modifications have been made to attenuate the virus (so, it could no longer cause herpes), to increase selectivity for cancer cells (so, it destroys cancer cells while leaving healthy cells unharmed), and to secrete the cytokine GM-CSF (a protein naturally secreted in the body to initiate an immune response). Inside a healthy cell, the modified virus is unable to replicate. Inside a cancer cell, the virus replicates and produces GM-CSF until cell lysis, releasing more virus, granulocyte-macrophage colony-stimulating factor (GM-CSF), and tumor antigens. GM-CSF attracts dendritic cells to the tumor site and activates them. These dendritic cells process and present the tumor antigen to T-cells. The T-cells then identify and destroy the cancer cells throughout the body. This approach is now in FDA phase II clinical trials.
Therapy directed to the tumor microenvironment
This therapeutic approach is based on the concept that tumor stem cells are protected from chemotherapy by their microenvironment. In the case of multiple myeloma, for example, the myeloma cells form junctions with the bone marrow stromal cells that support the interchange of signaling molecules that protect the myeloma cells. It was postulated that such junctions could be broken by the proteasome inhibitor bortezomib (PS-341; Velcade) which also prevents degradation of pro-apoptotic factors and thus allows programmed cell death.107,108 Treatment of multiple myeloma with combinations of bortezomib and thalidomide were approved by the FDA in 2008. In phase III trials, the combination of bortezomib and melphalan–prednisone is superior to melphalan–prednisolone in the treatment of older myeloma patients who were not eligible for high-dose chemotherapy; the complete response rate achieved was 32%. 109 In another study, bortezomib added to other treatment regimens significantly increased response rates.110,111 There are frequent side effects, such as neuropathy, fevers, hypoxia, and hypotension. Recently, a humanized monoclonal antibody (elotuzumab) to the signaling lymphocytic activation molecule F7 (SLAMF7) receptor has been developed. 112 This binds to the Fc receptor (CD-16), activates NK, and blocks interaction between SLAMF7 on myeloma cells and stromal cells. In preliminary trials, this has increased progression-free survival rates by 14% over lenalidomide plus dexamethasone therapy alone. 113 Significant side effects are also associated with this regimen, including fatigue, diarrhea, and fever.
Summary and conclusion
The immunotherapeutic approaches to cancer and their proved and projected results are listed in Table 2. The results of application of various cancer immunotherapeutics have repeatedly resulted in impressive anecdotal cures that cause optimism and stimulate hyperbolic press reports. Decreasing enthusiasm ensues as the results of controlled clinical trials are reported. Finally, an incremental result is obtained, usually limited to a relatively small number of patients with a highly specific type of cancer. In the case of Coley’s toxin, the first reported successful immunotherapy, no treatment using this approach currently remains. This is due to the inability to reproduce early anecdotal results and because of important and severe side effects (cytokine storm). Non-immune-specific approaches including BCG and IFN-α have resulted in successes in small groups of patients. Use of IL-2 and IL-2-expanded tumor-infiltrating lymphocytes was first reported to be a major breakthrough, but like Coley’s toxin did not stand the test of time. The use of adoptive cell transfer is now being tested using genetically engineered chimeric antigen receptor T-cells (CAR-T-cells) with the receptor linked to cell activation molecules. This approach is presently applied to a very small number of treatment-resistant B-cell tumors and is enormously expensive as it requires expansion and engineering of lymphocytes in vitro from each individual patient. In addition, it is associated with severe long-term side effects.
Summary of cancer immunotherapy approaches.

BCG: Bacille de Calmette de Guerin; DNCB: dinitrochlorobenzene; NK/LAK: natural killer cells/lymphokine activated killer cell; IFN-α: interferon-α; IL: interleukin; TILs: tumor-infiltrating lymphocytes; EGFR: epidermal growth factor receptor; HPV: human papillomavirus; HBV: hepatitis B virus; HCV: hepatitis C virus; CTL: cytotoxic T-lymphocyte.
In 1980s, engineered monoclonal antibodies attracted attention for the treatment of various cancers. Again, after extensive clinical trials, this treatment is now limited to certain kinds of cancer, for example, herceptin for EGF receptor-positive breast cancer and anti-CD-20 for B-cell tumors. Vaccines using tumor-specific transplantation-type antigens again have also shown promise for a few patients, but not as a general application. The most successful approach that has received less attention is the use of cancer virus vaccines. These have had an incredible role in preventing and even treating many cancers including liver, lymphoma, cervical, and other cancers. However, cancer causing viruses have not been identified for most human cancers. The latest immunotherapeutic emphasis is on checkpoint regulatory inhibitors. Several of these agents are FDA approved (an anti-CTLA-4 and two PD-1 antibodies) for specific oncologic conditions, and multiple additional CPRIs are under development. We are in the euphoric phase of the application of these therapies. There are many anecdotal cures that have been reported with this approach, but we are still in the mid-phase of larger clinical trials. This is a work in progress and just exactly how much overall impact this approach will have remains to be seen. However, the results so far are highly encouraging. These agents may work in a minority of cancer patients and are associated with severe and long-term side effects, but in those that respond this is clearly a welcome result. Hopefully, this approach will prove to be the exception to the rule of slight incremental improvement achieved with other immunotherapies for cancer.
Footnotes
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Dr Sell’s laboratory is supported by NIH grant CA161694.