
Abstract
The breast cancer susceptibility gene 1 (BRCA1) has been shown to maintain genomic stability through multiple functions in the regulation of DNA damage repair and transcription. Its translated BRCT (BRCA1 C-terminal domain) acts as a strong transcriptional activator. BRCA1 damaged by carboplatin treatment may lead to a loss of such functions. To address the possibility of the BRCA1 gene as a therapeutic target for carboplatin, we investigated the functional consequences of the 3′-terminal region of human BRCA1 following in vitro platination with carboplatin. A reduction in cellular BRCA1 repair of carboplatin-treated plasmid DNA, using a host cell reactivation assay, was dependent on the platination levels on the reporter gene. The transcriptional transactivation activity of the drug-modified BRCA1, assessed using a one-hybrid GAL4 transcriptional assay, was inversely proportional to the carboplatin doses. The data emphasized the potential of the BRCA1 gene to be a target for carboplatin treatment.
Introduction
The breast cancer susceptibility gene 1 (BRCA1) is a tumor gene located on chromosome 17q21. 1 The entire gene covers approximately 80 kb of the genomic sequence, and after identification it was subsequently cloned four years later. 2 The BRCA1 gene is composed of 24 exons, with an mRNA that is 7.8 kb in length, and 22 coding exons that translate into a protein of 1863 amino acids with a molecular weight of 220 kDa. 3 Its encoded protein has 3 major domains, including (1) an N-terminal RING finger domain (BRCA1 RING domain), (2) a large central segment with the nuclear localization signal (NLS), and (3) the BRCA1 C-terminal domain (BRCT). The BRCA1 protein plays an essential role in maintaining genomic stability associated with a number of cellular processes, including DNA repair, a cell cycle checkpoint, transcriptional regulation, and protein ubiquitination.4,5
The BRCA1 C-terminal domain (BRCT) contains two BRCT (BRCA1 C-terminal) domains in tandem (motif 1: amino acids 1653–1736, motif 2: amino acids 1760–1855). Each BRCT domain is characterized by a central, parallel four-stranded β-sheet with a pair of α-helices (α1 and α3) packed against one face, and a single α-helix (α2) packed against the opposite face of the sheet. 6 The two BRCA1-BRCT repeats interact in a head-to-tail fashion. This domain serves as a multipurpose protein–protein interaction module that binds to other BRCT repeats. 7 The BRCA1-BRCT domain has been identified as a phosphopeptide recognition module, and is demonstrated to bind to the phosphorylated protein partners (BACH1 and CtIP) known to participate in DNA damage repair and activation of transcription.8,9
Cancerous cells with inactivated BRCA1 have been reported to have defects in the repair of DNA double-strand breaks (DSBs).10,11 These cells have increased sensitivity to DNA-damaging agents that eventually result in major genomic instability and cell death. Carboplatin (Fig. 1), a second-generation platinum drug, is widely used for cancer treatment. 12 Its cytotoxicity to the cancer cells results from the formation of several types of platinum-DNA adducts 13 that interfere with DNA replication and transcription, and ultimately lead to the death of cancer cells. Carboplatin interacts nonspecifically with DNA and possibly with tumor suppressor genes in cancerous cells. Previously, in vitro interactions of the anticancer platinum drug carboplatin with the BRCA1 gene have been investigated. 12 The drug-modified BRCA1 stopped a cleavage by some restriction endonucleases, to imply that it preferentially attacked the 1,2-intrastrand d(GpG) crosslinks. Carboplatin was found to reduce the amount of amplified BRCA1 both in cells and cell-free systems. 12 In addition, its parental cisplatin was shown to modulate a repair-mediated transcriptional transactivation of the BRCA1 gene. 14 However, the functional consequence of the BRCA1 gene upon it being bound to carboplatin remains unexplored. Damage to intact BRCA1 by carboplatin might lead to an impairment of BRCA1 functions and ultimately cause cancer cell death. Here, we have described a detailed investigation of the functional analyses, host cell reactivation and transcriptional activation, of the specified DNA sequence of the 3′-terminal region of the BRCA1 gene induced by carboplatin.
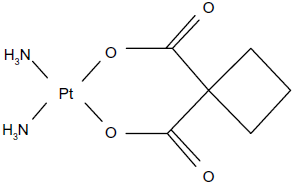
Chemical structure of carboplatin.
Materials and Methods
Cell culture
MCF-7 cells were purchased from the American Type Culture Collection (ATCC, Rockville, MD, USA), and grown in Dulbecco's modified Eagle's medium (DMEM) (Life Technologies, Paisley, UK) supplemented with 10% fetal bovine serum (FBS) and 1% penicillin–streptomycin. The cells were cultured at a constant temperature of 37 °C in a 5% carbon dioxide (CO,) humidified atmosphere.
Preparation of the 696-bp BRCA1 fragment
Messenger RNA (mRNA) was extracted from white blood cells using a mRNA isolation kit and biotinylated oligo(dT) (QIAGEN). The purified mRNA was used for complementary DNA (cDNA) synthesis, and the amplification of the 696-bp BRCA1 fragment (nucleotide 4,897–5,592) was performed with QIAGEN OneStep RT-PCR kit® (QIAGEN). 14 In brief, the RT-PCR mixture was prepared in a 1.5 mL microcentrifuge tube with a final volume of 50 μL, containing 5× reaction buffer, 400 μM of each dNTP, 0.6 μM forward primer (5′-AGCAGGGAGAAGCCAGAATTG-3′), 0.6 μM reverse primer (5′-TCAGTAGTGGCTGTGGGGGAT-3′), OneStep RT-PCR enzyme mix (QIAGEN), and RNase-free water. The template RNA was finally added to initiate the PCR reaction using a two-step thermal cycling. The first step comprised one cycle at 48 °C for 45 minutes, to allow for the synthesis of the first strand cDNA by the action of reverse transcriptase. The reverse transcriptase was inactivated at 94 °C for two minutes. The second step included 40 cycles of denaturation at 94 °C for 30 seconds, annealing at 60 °C for one minute and extension at 68 °C for two minutes. The extension of the final cycle was expanded to seven minutes. The amplified product was subjected to electrophoresis on a 1% agarose gel at 80 V for 60 minutes. The gel was stained with ethidium bromide, visualized under ultraviolet light, and the DNA extracted with a gel extraction kit (QIAGEN). The sequence of the purified 696-bp BRCA1 fragment was subsequently verified using an automated DNA sequencer (ABI PRISM™ 377, Perkin-Elmer, USA). The sequence data were aligned to the BRCA1 cDNA from GenBank (U-14680) using the SIM-4 program.
Construction of expression vector pBIND-BRCT
The sequence of the 696-bp BRCA1 fragment was double digested by restriction enzymes BamHI and XbaI. The cleaved 696-bp BRCA1 fragment was then cloned to the BamH1/XbaI site of the pBIND plasmid using T4 DNA ligase. The recombinant plasmid, termed pBIND-BRCT, was subsequently verified using an automated DNA sequencer (ABI PRISM™ 377, Perkin-Elmer, USA).
Cellular DNA repair
MCF-7 cells (1 × 106 cells) were incubated in a serum-free DMEM medium containing carboplatin (200 and 400 μM) at 37 °C for 24 hours. After drug exposure, the cells were washed twice with phosphate-buffered saline (PBS) and the medium was replaced by carboplatin-free DMEM. The genomic DNA was harvested at 4, 8, 16, and 24 hours and prepared. 12 The 3,426 bp fragment of BRCA1 exon 11 was then amplified using the isolated genomic DNA and specific forward (5′-GCCAGTTGGTTGATTTCCACC-3′) and reverse (5′-GTAAAATGTGCTCCCCAAAAGC-3′) primers. Product amplification was measured as described previously. 12
Host cell reactivation assay
The pBIND-BRCT or pBIND was incubated with carboplatin at various concentrations (0, 200, and 400 μM) at 37 °C for 24 hours. The platinated plasmid was precipitated with ethanol, washed, and co-transfected into MCF-7 cells by calcium phosphate precipitation along with the pSV-β-galactosidase, as an internal control. Cells were collected at various times after transfection. The expression level of the Renilla luciferase reporter gene was measured using a Luciferase® Reporter Assay System (Promega, USA).
Transcriptional transactivation of carboplatin-damaged BRCA1.
The pBIND-BRCT was incubated with carboplatin at various concentrations (0, 200, and 400 μM) at 37 °C for 24 hours. The platinated plasmid was precipitated with ethanol, washed, and co-transfected into MCF-7 cells by calcium phosphate precipitation along with the reporter plasmid pG5Luc and the pSV-β-galactosidase, as an internal control. The pG5Luc contained five GAL4-binding domain sites upstream of a minimal TATA box, which in turn was upstream of the firefly luciferase gene (luc+). Cell lysates were prepared and assayed for luciferase and β-galactosidase activities, respectively.
Results and Discussion
Construction of the expression vector pBIND-BRCT
The 3′-terminal region of the 696-bp BRCA1 fragment was cloned to the multicloning region of the pBIND at the BamHI and XbaI sites. The plasmid was transformed into Escherichia coli strain DH5α for multiplication and extracted by the alkaline lysis technique. 15 The recombinant plasmid was finally verified using an automated DNA sequencer (ABI PRISM™ 377, Perkin-Elmer, USA). DNA sequencing confirmed the recombinant pBIND-BRCT.
Cellular DNA repair
MCF-7 cells were treated with carboplatin at concentrations of 200 and 400 μM. After drug exposure at 37 °C for 24 hours, the 3,426-bp fragment of BRCA1 exon 11 was then amplified using isolated genomic DNA and specific forward and reverse primers. The results indicated a time-dependent recovery of carboplatin-modified genomic DNA (Fig. 2). A rapid recovery was observed at lower carboplatin concentration of 200 μM. However, more than 90% of the lesion repair was observed at both concentrations after 24 hours of repair time. The data showed the proficiency in repairing of MCF-7 cells on carboplatin treatment.
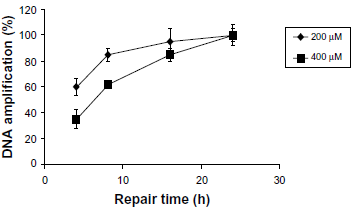
Proficiency in repairing carboplatin-damaged 3,426-bp BRCA1 exon 11 of MCF-7 cells. MCF-7 cells were incubated with medium plus carboplatin at concentrations of 200 and 400 μM at 37 °C for 24 hours. The cells were washed twice with PBS, and fresh medium was added. The genomic DNA was isolated at 4, 8, 16, and 24 hours and used as the template for the QPCR assay. 12 The data were derived from four independent experiments ± standard deviation (SD).
Cellular reactivation of carboplatin-treated BRCA1.
The transient expression of transfected DNA was used to study the effect of DNA damage on the transcriptional level and subsequently the ability of recipient cells to repair such lesions. The expression of the transfected genes depended on an intact coding sequence. Generally, a host cell reactivation assay is a technique used to study the proficiency of the host cells to repair a damaged plasmid. The drug-treated BRCA1 gene transfected into cells could not be expressed unless the recipient cells were given sufficient time to repair the lesions. In this study, the untreated pBIND-BRCT exhibited a maximal Renilla luciferase activity at 24 hours post-transfection (Fig. 3). After a prolonged transfection, a decrease in the Renilla luciferase activity was observed.
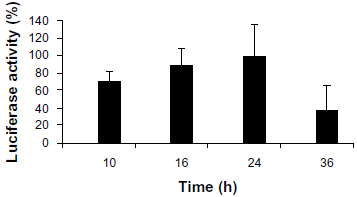
Time course of Renilla luciferase expression of pBIND-BRCT. DNA repair-proficient MCF-7 cells were transiently transfected with pBIND-BRCT along with the reporter plasmid pSV-β-galactosidase, as an internal control. Cell lysates were collected at 10, 16, 24, and 36 hours after transfection. The Renilla luciferase expression was detected using a Luciferase® Reporter Assay System. The data were derived from four independent experiments ± SD.
The Renilla luciferase activity of the carboplatin-treated pBIND-BRCT is shown in Figure 4. The results demonstrated that the cellular reactivation of platinated plasmid decreased as the carboplatin dose increased. It was notable that the Renilla luciferase activity at 400 μM decreased dramatically. The result indicated that a reduction in cellular reactivation of the drug-damaged Renilla luciferase encoding plasmid was a consequence of an increase in platination levels within the transcribed reporter gene. It is suggested that the platinum-BRCA1 adducts on the drug-treated pBIND-BRCT alter the DNA conformation that recruits DNA repair machinery and relevant proteins to sites of DNA damage.16,17 As a result, a sufficient time is required for cellular reactivation of such lesions to restore a normal nucleotide sequence of that specified BRCA1 gene. It has been demonstrated that the encoded product of the BRCA1 gene was involved in mediating the response to the chemotherapeutic treatment of cancer through homologous recombination (HR) during the repair of DNA DSBs. 18 Therefore, inactivation of the BRCA1 gene by the anticancer platinum drug carboplatin is possibly related to impaired BRCA1 functions normally responsible for repairing DNA adducts produced by the drug, and ultimately resulted in cancer cell death.
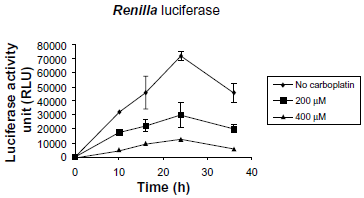
Time course of Renilla luciferase expression of pBIND-BRCT. The pBIND-BRCT or pBIND was incubated with carboplatin at concentrations of 0, 200, and 400 μM at 37 °C for 24 hours. Platinated plasmid was precipitated by ethanol, washed, and co-transfected into MCF-7 cells along with the pSV-β-galactosidase, as an internal control. Cells were collected at various times after transfection. The expression level of the Renilla luciferase reporter gene was measured using a Luciferase® Reporter Assay System (Promega, USA). The data were derived from four independent experiments ± SD.
Effect of carboplatin on the transcriptional transactivation of the 3′-terminus of BRCA1.
In a previous experiment, it was demonstrated that the BRCT domain (amino acids 1528–1863) stimulated gene transcription in transiently transfected human cells. 19 It has been reported that the GAL4–BRCA1 plasmid transactivated a reporter template bearing four GAL4-binding sites upstream of the c-fos minimal promoter fused to the luciferase gene. This system has been used to study the effects of mutation on the BRCT domain, and the interaction of BRCT domain with other proteins.20–22
The pBIND plasmid, a high copy number plasmid in which the human cytomegalovirus (CMV) immediate early promoter drives the expression of the yeast GAL4 gene that codes for a portion of the protein (amino acids 1–147) containing a DNA-binding domain, has been used in a “two hybrid assay” system for studying the interaction of any two proteins.19,23 In our modified “one-hybrid assay” system, the BRCT domain was fused to the GAL4 protein in the pBIND plasmid (Fig. 5). This fusion protein also increased the firefly luciferase expression over the negative controls. The transcriptional transactivation activity was observed when the BRCT domain was constructed in the pBIND plasmid.

Schematic expression of the GAL4-BRCT from the pBIND-BRCT plasmid. The GAL4-BRCT protein activated the transcription of the firefly luciferase reporter gene on the pG5 Luc plasmid.
To investigate whether the drug-treated BRCA1 was able to transactivate the expression of the firefly luciferase gene, the DNA repair-proficient MCF-7 cells were transiently transfected with the carboplatin-treated pBIND-BRCT along with the reporter plasmid pG5Luc. The pBIND plasmid, harboring only the GAL4 DNA-binding protein that is unable to activate the expression of firefly luciferase from the pG5Luc plasmid, was used as a negative control. The pBIND-BRCT was platinated with carboplatin at a concentration of 0, 200, and 400 μM at 37 °C for 24 hours. The platinated plasmid was precipitated by absolute ethanol and dissolved in double distilled water, and co-transfected with the pG5Luc and the pSV-β-galactosidase (as an internal control) in the MCF-7 cells. Cell lysates were prepared at 10, 16, 24, and 36 hours after transfection. The firefly luciferase activity was measured using a Dual-Luciferase® Reporter Assay System. The data were derived from four independent experiments ± SD. After 10, 16, 24, and 36 hours of transfection, the firefly and Renilla luciferase activities were measured. The firefly luciferase activity was significantly decreased at a carboplatin concentration of 400 μM (Fig. 6). A decrease in the firefly luciferase activity was observed after prolonged transfection.
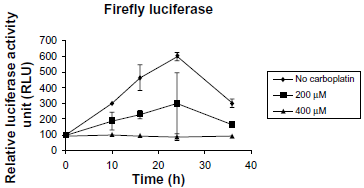
Time course of firefly luciferase expression. The pBIND-BRCT was incubated with carboplatin at concentrations of 0, 200, and 400 μM at 37 °C for 24 hours. The platinated plasmid was precipitated with ethanol, washed, and then co-transfected into MCF-7 cells with the pG5 Luc and the pSV-β-galactosidase, as an internal control. A cell lysate was prepared at 10, 16, 24, and 36 hours after transfection. The firefly luciferase expression was detected by a Dual-Luciferase® Reporter Assay System. The data were derived from four independent experiments ± SD.
It was noted that the ability of the cellular reactivation and transcriptional activation of the drug-treated BRCA1 occurred in a dose–response manner. These data agree very well with our previous study of in vitro inhibition of BRCA1 amplification induced by carboplatin. 12 Therefore, it is suggested that a decrease in transcriptional transactivation was mediated by the cellular repair ability and the transcriptional regulation of the carboplatin-damaged BRCA1. It is likely that carboplatin exerted its inhibitory effect through distortion of the DNA base pairs of the Pt-DNA adducts that interfered with the RNA polymerase II as well as the promoters.24–28 It has been reported that the BRCA1–RNA polymerase II complex was capable of activating the p21 promoter and stabilizing p53 in response to DNA damage.29,30 The interaction of BRCA1 and the p53 potentially resulted in the redirection of a p53-mediated transactivation from the apoptotic target genes involved in DNA repair and cell cycle arrest. 30 In addition, Pt-DNA adducts may inhibit transcription initiation by hijacking transcription factors that bind to the Pt-DNA adducts.24,25 Furthermore, the Pt-DNA adducts can disrupt chromatin remodeling/nucleosome mobility. 24
Recently, the emerging highlighted therapeutic strategies appear to be targeting the DNA damage response pathways.10,11,31 A newer approach to the use of platinum agents, cisplatin, and carboplatin to treat triple-negative breast cancer (TNBC) patients is currently being assessed in clinical trials on the basis of the dysfunction of BRCA1. 32 The decreased BRCA1 expression in TNBC sensitizes patients to cisplatin occurring with an improved efficacy.33,34 Carboplatin has recently been used in dual combination chemotherapy with docetaxel among TNBC patients. 35 In addition, the combined regimen of carboplatin and TRA-8, a monomeric monoclonal antibody, was found to improve the therapeutic efficacy in the treatment of ovarian cancer. 36 Moreover, such a combination potentiated therapeutic responses among TNBC patients and would be beneficial to assess any early tumor responses to therapy. 37 Together, knowing the status of the DNA repair pathways such as a loss or defect of the BRCA1 gene could predict TNBC patients’ sensitivity to carboplatin-based chemotherapy.38,39
Author Contributions
Conceived and designed the experiments: AR. Analyzed the data: AR. Wrote the first draft of the manuscript: AR. Agree with manuscript results and conclusions: AR, BC. Jointly developed the structure and arguments for the paper: AR, BC. Made critical revisions and approved final version: AR, BC. All authors reviewed and approved of the final manuscript.
Disclosures and Ethics
This manuscript has been read and approved by all authors. The authors have confirmed that the published article is unique and not under consideration nor published by any other publication and that they have permission to reproduce any copy-righted material. The authors declare no conflicts of interest.
Footnotes
Acknowledgments
We are grateful to Dr. Brian Hodgson for assistance in English, and the Pharmaceutical Laboratory Service Center, Faculty of Pharmaceutical Sciences, Prince of Songkla University for research facilities.