
Abstract
The transcription factor TWIST has been reported to play an important role in tumor progression as well as resistance to anti-cancer drugs. However, the role of TWIST in gastric cancer and the molecular mechanisms by which this protein elicits drug resistance remain poorly understood. We transfected gastric cancer cell lines with lentiviral vector to generate TWIST-overexpressing stable cell lines. Our study showed that overexpression of TWIST not only increased cell migration and invasion but also induced resistance to the anti-cancer drug paclitaxel in gastric cancer. Paclitaxel increased gastric cancer cell death in dose-dependent manner; this was decreased following TWIST overexpression. Furthermore, treatment with paclitaxel decreased Akt phosphorylation and Bcl-2 expression, whereas these effects were suppressed by TWIST overexpression. Treatment of cells with Akt inhibitor or small interfering RNA targeting for Bcl-2 led to increased paclitaxel-induced cell death, indicating that TWIST elicits resistance to paclitaxel via the regulation of the Akt and Bcl-2 pathway. Our results suggest an underlying mechanism for TWIST-mediated paclitaxel resistance and indicate that TWIST represents a potential target for overcoming paclitaxel resistance in gastric cancer cells.
Introduction
TWIST is a well-known key transcription factor that promotes epithelial–mesenchymal transition (EMT). 1 Elevated TWIST expression results in the inhibition of E-cadherin expression and the promotion of N-cadherin expression, which activates EMT, thereby contributing to invasion and metastasis of cancer cells. 2 Therefore, TWIST has been suggested to act as an oncogene. Overexpression of TWIST has been observed in a variety of cancers, including prostate, lung, breast, and gastric cancer and is reported to be associated with invasive pathological subtypes and poor prognosis.3–7
In addition, TWIST triggers resistance or decreases sensitivity to anti-cancer drugs. High TWIST expression promotes resistance to paclitaxel in nasopharyngeal carcinoma cells 8 and to Imatinib in chronic myeloid leukemia. 9 In breast cancer cells, overexpression of TWIST promotes multidrug resistance.10,11 Most cytotoxic anti-cancer drugs have been shown to elicit the apoptosis of tumor cells and decrease TWIST expression, while TWIST has been reported to protect cells from anti-cancer drug-induced apoptosis. TWIST overexpression is associated with resistance to anti-cancer drugs via the inhibition of apoptosis in colon cancer cells and laryngeal carcinoma.12,13
However, the role of TWIST in eliciting drug resistance in gastric cancer and the underlying mechanism remain to be elucidated. Previous studies have shown that TWIST expression is upregulated in gastric cancer with poor outcomes.6,7 Moreover, this transcription factor is suggested to promote the migration, invasion, and metastasis of gastric cancer cells. Therefore, in this study, the effect of TWIST overexpression on resistance to anti-cancer drugs in two gastric cancer cell lines and the underlying mechanism were investigated. We found that TWIST overexpression induced resistance to paclitaxel by inhibiting apoptosis and Akt and Bcl-2 were important mediator of paclitaxel resistance in gastric cancer cells.
Methods and materials
Reagents
Paclitaxel, fibronectin, Hoechst 33258, and 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) were purchased from Sigma-Aldrich Chemical (USA). LY294002 was obtained from Calbiochem (USA). Antibodies were obtained from Cell Signaling Technology, Inc. (USA). All other chemicals were of the highest commercial grade available.
Cell culture
SNU216 and SNU484 cells were obtained from the Korean Cell Line Bank (Korea) and were maintained. The cells were grown in RPMI 1640 (Gibco, Thermo Fisher Scientific Inc., USA) containing 10% fetal bovine serum (FBS; Gibco) at 37°C in 95% air/5% CO2 incubator. When the cultures reached confluence, subculture was prepared using a 0.02% ethylenediaminetetraacetic acid (EDTA)–0.05% trypsin solution. The cells were grown on tissue culture plates in RPMI 1640 medium containing 10% FBS. Serum was removed from culture media at the time of agent addition.
Lentiviral-based overexpression of TWIST
Lentiviral-based overexpression was used for overexpression of TWIST. A pLKO lentiviral vector targeting TWIST or an empty pLKO vector was used for overexpression of TWIST in the SNU216 and SNU484 cells. Lentivirus stocks were produced using the ViraPowerTM Lentiviral Packaging Mix and the 293FT cell line according to the manufacturer’s protocol (Invitrogen, USA). SNU216 and SNU484 cells grown to 50% confluence were incubated for 24 h in a 1:1 dilution of virus:media with 5 µg/mL Polybrene. After a 24-h recovery period in complete media without virus, polyclonal stable cell lines were selected and maintained in media containing 5 µg/mL puromycin. Overexpression of TWIST was determined by reverse transcription polymerase chain reaction (RT-PCR), western blotting, and immunocytochemistry.
Knockdown of Bcl-2 using small interfering RNA
Cells were transfected with multiple small interfering RNA (siRNA) using Lipofectamine RNAiMAX reagent (Invitrogen, Life Technologies Corporation, USA) according to the manufacturer’s instructions. The sequences of multiple siRNA are as follows: Bcl-2 siRNA (Dharmacon), 5′-GGGAGAACAGGGUACGAUA-3′, 5′-GAAGUACAUCCAUUAUAAG-3′, 5′-GGAGGAUUGUGGCCUUCUU′, and 5′-UCGCCGAGAUGUCCAGCCA-3′. RNAi negative control was obtained from Invitrogen.
Measurement of cell viability
Cell viability was evaluated using an MTT assay. After washing the cells, culture medium containing 0.5 mg/mL of MTT was added to each well. The cells were incubated for 2 h at 37°C, the supernatant was removed, and the formed formazan crystals in viable cells were solubilized with 0.11 mL of dimethylsulfoxide. A 0.1 mL aliquot of each sample was then transferred to 96-well plates and the absorbance of each well was measured at 570 nm with spectrophotometer (Hewlett-Packard, Agilent Technologies, USA). Data were expressed as a percentage of control measured in the absence of paclitaxel.
Cell-cycle analysis
Cells were grown in six-well plates and were treated as indicated. Then, attached and floating cells were pooled, pelleted by centrifugation, washed in phosphate-buffered saline (PBS), and fixed with cold 70% ethanol containing 0.5% Tween 20 at 4°C overnight. Cells were washed and resuspended in 1.0 mL of propidium iodide solution containing 100 μg of RNase A/mL and 50 μg propidium iodide/mL and incubated for 30 min at 37°C. Cells were assayed using FACSAria (BD Biosciences, USA) at 488 nm and data were analyzed. Cells with sub-G1 propidium iodide incorporation were considered as apoptotic. The percentage of apoptotic cells was calculated as the ratio of events on sub-G1 to events from the whole population.
Cell migration and invasion assays
Gastric cancer cells were harvested with 0.05% trypsin containing 0.02% EDTA (Sigma-Aldrich) and suspended in RPMI medium. For the migration assay, membrane filters (8-μm pore size) in disposable 96-well chemotaxis chambers (Neuro Probe, USA) were pre-coated with 5 mg/mL fibronectin for 4 h at room temperature. Cells (3 × 103 cells/well) were loaded into the upper chambers, and 1% FBS was loaded into the lower chamber. After 24 h of incubation, non-migrating cells were removed from the upper chamber with a cotton swab, and the cells on the lower surface of the insert were stained with Hoechst 33342 (Sigma-Aldrich). Migrated cells were counted under a fluorescence microscope at 10× magnification.
For the invasion assay, 3 × 104 cells/well were seeded in the upper chamber, which was coated with 30 μL of Matrigel (1 mg/mL in cold medium; BD Transduction Laboratories, USA). Serum-free medium containing 1% FBS or control vehicle was added to the lower chamber. After 24 h of incubation, non-invading cells were removed from the upper chamber with a cotton swab, and cells on the lower surface of the insert were stained with Hoechst 33342 (Sigma-Aldrich). Invasive cells were counted under a fluorescence microscope at 10× magnification.
Real-time RT-PCR
Total RNA was extracted from gastric cancer cells, using TRIzol reagent (Invitrogen, Life Technologies Corporation) following the manufacturer’s instructions. An amount of 2 μg of RNA was reverse transcribed with SuperScript (Invitrogen) in a final volume of 20 μL. The 2 μL of complementary DNA (cDNA) was amplified with each primer and SYBRGreen (Applied Biosystems, Life Technologies, USA) using the fluorescence reader Corbett Rotor-Gene 6000 (QIAGEN, Inc., USA). The primers used were the following: glyceraldehyde 3-phosphate dehydrogenase (GAPDH) 5′-TCCATGACAACTTTGGTATCG-3′, 5′-TGTAGCCAAATTCGTTGTCA-3′; TWIST 5′-AGTCTTACGAGGAGCTGC-3′, 5′-CGGCCAGGTACATCGACT-3′. The number of cycles in the PCR was determined for each gene and ranged from 25 to 35. Data were normalized to GAPDH, and messenger RNA (mRNA) abundance was calculated using the 2−ΔΔCT method. The PCR products were also confirmed by mobility on gel electrophoresis.
Western blotting
Cells were harvested at various times after paclitaxel treatment and disrupted in lysis buffer (1% Triton X-100, 1 mM ethyleneglycol-bis(2-aminoethyl ether)-N,N,N′,N′-tetraacetic acid (EGTA), 1 mM EDTA, 10 mM Tris-HCl, pH 7.4, and protease inhibitors). Cell debris was removed by centrifugation at 10,000g for 10 min at 4°C. The resulting supernatants were resolved using sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) and transferred onto nitrocellulose membranes. The membranes were blocked with 5% non-fat dried milk at room temperature for 30 min and incubated with anti-TWIST (Bio Matrix Research, Japan), anti-phospho Akt (Cell Signaling Technology, Inc.), anti-Bcl-2 (Epitomics, USA), and anti-GAPDH. The membranes were then washed and incubated with horseradish peroxidase–conjugated secondary antibody. Signals were visualized using enhanced chemiluminescence (Amersham, UK). All western blot densitometry data were normalized for the loading control GAPDH by CS Analyzer.
Immunocytochemistry
Cells overexpressing TWIST were cultured on glass coverslips. Cells were washed twice with PBS, fixed with 4% paraformaldehyde in PBS for 10 min, and permeabilized with 0.5% Triton X-100 in PBS for 10 min. After washing twice with PBS, cells were blocked with 8% bovine serum albumin (BSA) in Tris-buffered saline Triton X-100 (TBST). Cells were incubated with rabbit polyclonal anti-AIF overnight 4°C and washed twice with TBST. Cells were incubated with fluorescein isothiocyanate (FITC)-conjugated secondary antibody (Jackson ImmunoResearch Laboratories, USA) for 1 h, and the nuclei were counterstained with 4′,6-diamidino-2-phenylindole (DAPI). Cells were washed twice and visualized using the confocal microscope.
Statistical analysis
The data are expressed as mean ± standard error of the mean (SEM) and the difference between two groups was evaluated by unpaired Student’s t-test using SPSS v10.1 (SPSS Inc., USA). A probability level of 0.05 was used to establish significance.
Results
Overexpression of TWIST in gastric cancer cells
In order to investigate the function of TWIST in gastric cancer, gastric cancer cell lines SNU216 and SNU484 were transfected with a TWIST1-expressing lentiviral vector. Following transfection, TWIST1 mRNA levels were found to be increased in these cells (Figure 1(a)). The protein expression levels of TWIST were additionally confirmed by western blot analysis and immunocytochemistry. Transfection with the lentiviral vector increased the expression of TWIST protein; however, the expression of this protein was not detected in cells transfected with empty vector (Figure 1(b) and (c)).

Overexpression of TWIST. Gastric cancer cell lines, SNU216 and SNU484, were infected with lentiviruses expressing either the empty pLKO vector (EV) or TWIST to generate stable TWIST-overexpressing cells. The overexpression of TWIST was detected by (a) qRT-PCR, (b) western blotting, and (c) immunocytochemistry. (a) TWIST1 gene expression was normalized by GAPDH expression.
Overexpression of TWIST promotes migration and invasion of gastric cancer cells
We investigated whether TWIST is involved in tumorigenesis as well as in migration and invasion of cells in gastric cancer. Our findings revealed that TWIST overexpression significantly elevated migration and invasion in SNU216 and SNU484 cell lines, indicating that this transcription factor is required for the migration and invasion of gastric cancer cells (Figure 2(a)–(d)).

Role of TWIST in migration and invasion of gastric cancer cells. Migration and invasion assays were performed using empty vector (EV) or TWIST-overexpressing stable cells (TWIST). Overexpression of TWIST enhanced (a and b) migration and (c and d) invasion in the presence of 1% FBS. The graph shows the number of cells that migrated and invaded in the presence of 1% FBS or under serum-free (S/F) conditions. Representative data show the cells that migrated and invaded in the presence of 1% FBS.
Overexpression of TWIST enhances drug resistance in gastric cancer cells
In order to investigate whether high TWIST expression causes resistance to anti-cancer drugs, SNU216 and SNU484 gastric cancer cell lines were exposed to various concentrations of paclitaxel for 24 h. Paclitaxel treatment induced loss of cell viability in a dose-dependent manner. However, TWIST overexpression suppressed paclitaxel-induced cell death (Figure 3(a) and (b)). Furthermore, we analyzed the cell cycle to verify whether paclitaxel-induced cell death resulted from apoptosis and the effects of TWIST. DNA content below that of cells in the G0/G1 phase suggested cell death by apoptosis. 14 Treatment with paclitaxel resulted in the accumulation of Sub G0/G1 DNA content in empty vector–transfected gastric cancer cells, while overexpression of TWIST reduced paclitaxel-induced apoptosis (Figure 3(c) and (d)). These results indicate that paclitaxel induces apoptosis in gastric cancer cells and that high levels of TWIST may exert anti-apoptotic effects, resulting in resistance to this drug.

TWIST-induced resistance to paclitaxel in gastric cancer cells. (a and b) Cells were treated with paclitaxel at the indicated concentration for 24 h and cell death was measured by MTT assay. (c and d) Apoptosis was measured by cell-cycle analysis; data show the percentage of cells in sub-G1 phase. Data are means ± SEM of four independent experiments (*p < 0.05 compared with empty vector–expressing cells).
Overexpression of TWIST promotes drug resistance by regulating Akt and Bcl-2
In order to elucidate the mechanisms by which TWIST induces paclitaxel resistance, we investigated the expression of Akt and Bcl-2. Akt pathways are crucial in regulating cell survival in cancer cells. 15 Akt phosphorylation and Bcl-2 expression were found to be elevated in cells overexpressing TWIST. Treatment with paclitaxel decreased Akt phosphorylation and Bcl-2 expression, which occurred at higher levels in TWIST-overexpressing cells than in empty vector–transfected cells (Figure 4). These results indicate that the Akt pathway and Bcl-2 are involved in the TWIST-mediated response to paclitaxel in gastric cancer cells.
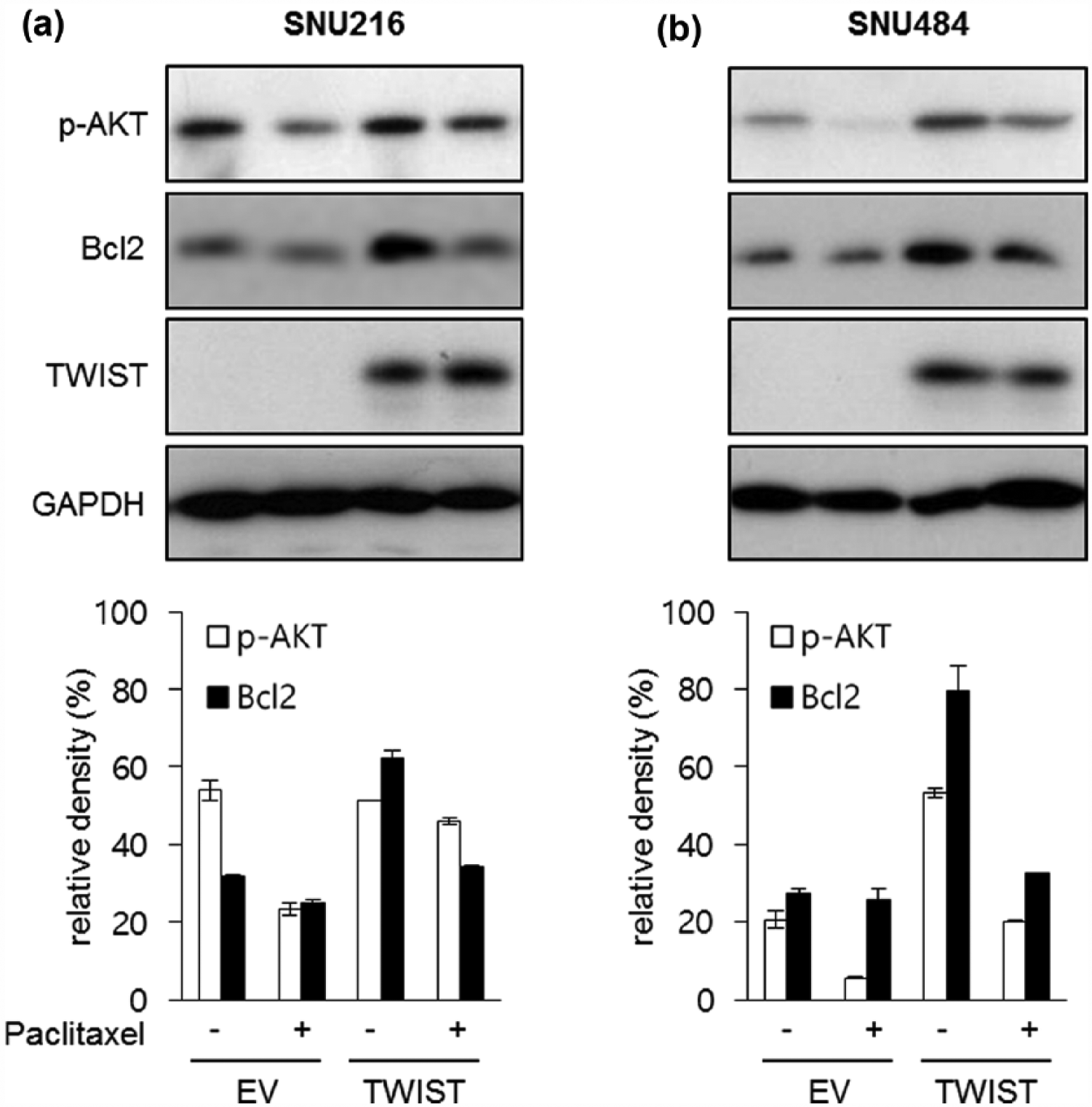
Overexpression of TWIST increases Akt activation and Bcl-2 expression. Akt phosphorylation and Bcl-2 expression were analyzed after exposure to paclitaxel (50 ng/mL) in empty vector (EV) or TWIST-overexpressing cells (TWIST). Western blot and densitometry analyses in (a) SNU216 and (b) SNU484 indicated that increased Akt phosphorylation and Bcl-2 expression were observed in TWIST-overexpressing cells after paclitaxel treatment.
We further confirmed the involvement of Akt and Bcl-2 in TWIST-mediated paclitaxel resistance. TWIST-induced levels of Akt phosphorylation were lower in cells treated with both paclitaxel and LY294002, which led Akt phosphorylation repressed via inhibition of PI3K, than in cells treated with paclitaxel alone (Figure 5(a)). Inhibition of Akt decreased paclitaxel-induced cell viability and increased apoptosis (Figure 5(b) and (c)). In addition, siRNA targeting Bcl-2 decreased TWIST overexpression-induced Bcl-2 expression (Figure 6(a)). The inhibition of Bcl-2 additionally increased paclitaxel-induced cell death and apoptosis (Figure 6(b) and (c)). These results show that the inhibition of Akt and Bcl-2 induced increased sensitivity to paclitaxel in TWIST-overexpressing cells, suggesting that Akt and Bcl-2 may play a key role in TWIST-mediated paclitaxel resistance in gastric cancer cells.

Effect of Akt on TWIST-mediated paclitaxel resistance. Cells were treated with paclitaxel (50 ng/mL) in the presence of the Akt inhibitor LY294002 (10 μM) or not. Inhibition of Akt led to decreased (a and b) paclitaxel-induced Akt phosphorylation and increased (c) paclitaxel-induced cell death and (d) apoptosis in TWIST-transfected cells. Data are mean ± SEM of three independent experiments (a and b: *p < 0.05 vs paclitaxel– LY294002+ group; c and d: *p < 0.05 vs paclitaxel group).

Effect of Bcl-2 on TWIST-mediated paclitaxel resistance. Cells were treated with paclitaxel (50 ng/mL) in the presence of siRNA mixture targeting Bcl-2 or not. Inhibition of Bcl-2 resulted in a decrease in (a and b) paclitaxel-induced Bcl-2 expression and increase in (c) paclitaxel-induced cell death and (d) apoptosis in TWIST-transfected cells. Data are mean ± SEM of three independent experiments (a and b: *p < 0.05 vs paclitaxel– LY294002+ group; c and d: *p < 0.05 vs paclitaxel group).
Discussion
TWIST is overexpressed in various cancers such as those of the breast, 3 prostate,15–17 lung, 5 and liver 18 as well as in gastric carcinoma.18,19 This transcription factor has been shown to play an essential role in tumor progression. TWIST activates the EMT process, which contributes to invasion and metastasis. 20 In addition, the overexpression of TWIST is associated with poor survival in cancer patients and increased aggressiveness of disease.21–23 TWIST has been recently shown to promote migration and invasion in gastric cancer cells.6,19 Yang et al. 19 have reported that TWIST elicits an increase in N-cadherin and fibronectin levels, which promotes cell migration and invasion. The present findings also confirm that overexpression of TWIST increases migration and invasion of gastric cancer cells.
Furthermore, TWIST has reported to inhibit apoptosis in cancer cell and induced G2 cell-cycle arrest24–26 and cause drug resistance in cancer cells.13,27,28 However, it remains unknown whether TWIST elicits resistance to anti-cancer drugs in gastric cancer cells. The taxane paclitaxel is an antimitotic agent that induces cell-cycle arrest at the G2/M stage as well as apoptosis in cancer cells. 29 This agent has been used to treat advanced gastric cancer, and the weekly administration of paclitaxel has been found to be effective in several types of cancer.30,31 Although paclitaxel is one of the most effective anti-cancer drugs, resistance is often acquired by cancer cells. Therefore, an understanding of the mechanisms involved in resistance to paclitaxel is necessary in order to improve the efficacy of anti-cancer therapies. In this study, paclitaxel was found to induce apoptosis in gastric cancer cells; however, TWIST overexpression suppressed paclitaxel-induced apoptosis, suggesting that the level of TWIST expression may influence the response to paclitaxel. In addition, TWIST overexpression was found to trigger resistance to paclitaxel via the regulation of Akt and Bcl-2 expression.
TWIST has been shown to activate the Akt signaling pathway in several types of cancer.10,32 Cheng et al. 10 have reported that TWIST promotes migration and invasion of breast cancer cells and induces resistance to paclitaxel via the activation of Akt pathway and β-catenin. Moreover, it has been reported that these activities are suppressed by the silencing of Akt. 10 In ovarian cancer, TWIST has been shown to contribute to cisplatin resistance via the Akt pathway. 33 TWIST-mediated taxol resistance is also associated with Akt expression in nasopharyngeal carcinoma cells. 34 Consistent with these results, we found that high TWIST expression increased Akt activation and suppressed paclitaxel-induced apoptosis. Moreover, inactivation of Akt via an Akt inhibitor, LY294002, increased sensitivity to paclitaxel. Our results indicate that TWIST-mediated paclitaxel resistance may be regulated through the Akt pathway, which has been considered as a therapeutic target in cancer. 15 Therefore, our results also implicate Akt as a target for TWIST-mediated paclitaxel resistance.
It has been reported that taxanes interact with Bcl-2. 35 In this study, paclitaxel may be considered to have decreased Bcl-2 expression through direct binding, resulting in apoptosis. However, TWIST overexpression was found to increase Bcl-2 expression. In addition, transfection of siRNA targeting Bcl-2 increased paclitaxel-induced cell death and apoptosis. TWIST has been reported to exert oncogenic properties by regulating Bcl-2 in gastric cancer cells. 36 Patients with hepatocellular carcinoma that coexpresses Bcl-2 and TWIST have been shown to exhibit poor prognosis. 18 These reports support our suggestion that Bcl-2 plays an important role in TWIST-mediated paclitaxel resistance. Moreover, it has been reported that TWIST promotes survival of cancer cells exposed to anti-cancer drugs through reduction of pro-apoptotic factors Bak and Bad expression.34,37 These results suggest that TWIST may mediate resistance of cancer cells via regulation of anti- as well as pro-apoptotic factors. It remains to be clarified whether Bak and Bad are related with TWIST-mediated paclitaxel resistance of gastric cancer cells in the further study.
In summary, our findings provide novel insights into the role of TWIST in the progression of gastric cancer as well as elucidate the mechanism underlying TWIST-mediated paclitaxel resistance. Our results suggest that TWIST may serve as a predictor for paclitaxel resistance in gastric cancer. In addition, our results suggest that the Akt pathway and Bcl-2 represent targets for overcoming TWIST-mediated paclitaxel resistance in gastric cancer cells.
Footnotes
Acknowledgements
D.Y.P. and C.H.K. conceived and designed the experiments; C.H.K., H.J.P., Y.C., Y.J.W., and S.J.L. performed and validated the experiments; H.J.P. and Y.C. analyzed the data; D.Y.P., C.H.K., and Y.C. wrote the paper; D.Y.P. involved in supervision; D.Y.P. obtained funding; and C.H.K., H.J.P., and Y.C. contributed equally to this study.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was supported by a grant (No. 0920050) from the National Research and Development Program for Cancer Control, Ministry for Health, Welfare and Family Affairs, Republic of Korea.