
Abstract
Epstein–Barr virus is a ubiquitous virus and is associated with several human malignances, including the significant subset of gastric carcinoma, Epstein–Barr virus–associated gastric carcinoma. Some Epstein–Barr virus–associated diseases are uniquely prevalent in populations with different geographic origins. However, the features of the disease and geographically associated Epstein–Barr virus genetic variation as well as the roles that the variation plays in carcinogenesis and evolution remain unclear. Therefore, in this study, we sequenced 95 geographically distinct Epstein–Barr virus isolates from Epstein–Barr virus–associated gastric carcinoma biopsies and saliva of healthy donors to detect variants and genes associated with gastric carcinoma and population structure from a genome-wide spectrum. We demonstrated that Epstein–Barr virus revealed the population structure between North China and South China. In addition, we observed population stratification between Epstein–Barr virus strains from gastric carcinoma and healthy controls, indicating that certain Epstein–Barr virus subtypes are associated with different gastric carcinoma risks. We identified that the BRLF1, BBRF3, and BBLF2/BBLF3 genes had significant associations with gastric carcinoma. LMP1 and BNLF2a genes were strongly geographically associated genes in Epstein–Barr virus. Our study provides insights into the genetic basis of oncogenic Epstein–Barr virus for gastric carcinoma, and the genetic variants associated with gastric carcinoma can serve as biomarkers for oncogenic Epstein–Barr virus.
Keywords
Introduction
Gastric carcinoma is the third leading cause of cancer mortality, with 723,000 deaths worldwide. 1 Many gastric carcinomas are associated with infection by Helicobacter pylori and Epstein–Barr virus (EBV). EBV-associated gastric carcinoma (EBVaGC) comprises approximately 10% of all cases of gastric carcinoma.2–6 EBV, also called human herpesvirus 4 (HHV-4), is a ubiquitous virus with an infection rate of approximately 90% of the worldwide adult population. 7 EBV is associated with multiple lymphoproliferative diseases, including infectious mononucleosis, Burkitt’s lymphoma, natural killer (NK)/T-cell lymphoma, diffuse large B-cell lymphoma, and post-transplant lymphoproliferative disorders, as well as epithelial carcinomas, including nasopharyngeal carcinoma and gastric carcinoma.
Due to the important roles of EBV in so many malignancies, previous research has elucidated the crucial pathogenesis of the virus genome and genes and provided many insights into understanding viral infection and pathogenesis. The oncogenic effects of EBV nuclear antigens (EBNAs) and latent membrane proteins (LMPs) were validated through the interaction with some tumor suppressor genes and signaling pathways in EBV-associated cancers.8–11 Furthermore, the genomic variations in viral genes may affect latency maintenance and immune recognition during persistent infection. 12 EBVaGC expresses EBNA1, EBERs, BARTs, and LMP2A as the EBV latency I tumor, 13 and several viral genes induce the promoter methylation of host genes to contribute to the development of gastric cancer.14–16 However, the precise atlas of genomic variations involved in gastric cancer carcinogenesis remains undetermined.3,4,6,17
Although the EBV-related malignancies nasopharyngeal carcinoma and Burkitt’s lymphoma have notably different incidence rates in different areas throughout the globe, EBV infection accounts for 6%–10% of gastric carcinomas without regional or racial differences.2,3,5 Two questions remain to be elucidated with regard to EBV genetics: first, whether EBV strains have genetic structures among populations, and second, whether certain EBV subtypes are associated with elevated disease risk. Therefore, virus sequence variation requires additional study. The characterization of fragmented and regional variation of the viral genome, small sample size, and restricted sample origins have limited a comprehensive understanding of genome variants and their importance in virus distribution, evolution, and oncogenesis.
In this study, by the approaches of virus-targeted capture and next-generation sequencing, we performed a genome-wide analysis and a case-control study to uncover the EBV genes associated with gastric carcinoma that might contribute to the oncogenesis of EBVaGC and genes associated with the geographic difference.
Results
Targeted sequencing and variation analysis of EBV genomes
We performed whole genome sequencing of EBV from 41 specimens isolated from patients diagnosed with gastric carcinoma and 54 saliva samples from healthy donors. Of them, 30 and 65 samples, respectively, had geographic origins of North China and South China. The sample characteristics were summarized in Table 1. The sequencing depths varied from 19-fold to 9339-fold, with an average of 1523-fold, and the target region coverage mapped to the reference genome EBV-WT (NC007605.1) ranged from 93.44% to 99.91%, with an average of 98.08%. Details of sample information and sequencing data are shown in Table S1 and Table S2, respectively. The good quality of sequencing made a solid base for subsequent analyses.
Sample characteristics of the EBV GWAS.

EBV: Epstein–Barr virus; GWAS: Genome-wide association study.
The p value was calculated by two-sided Pearson chi-square or Fisher’s exact test.
In these EBV sequences, 6209 variants were identified in comparison to the reference EBV-WT, including 5770 substitutions, 138 insertions, and 212 deletions (Table S3). To discover the most diverse parts of the EBV genome, the number of variants was calculated within a 1000-base-pair sliding window. We observed that the latent genes were the highest variable part of the genome and the lytic genes had a lower variation frequency (Figure 1). This result was similar to previous studies.12,18 Moreover, in these variants, based on the categories of defined function of virus proteins, 19 non-synonymous variants were harbored abundantly in latency, tegument, and membrane protein genes, such as EBNA3A, BPLF1, EBNA3C, EBNA3B, and LMP1, whereas the genes with roles in nucleotide metabolism and packaging harbored few non-synonymous variants (Figure 2, Table S4). The details of the variations in individual genomes are summarized in Table S3 in the supplemental material.
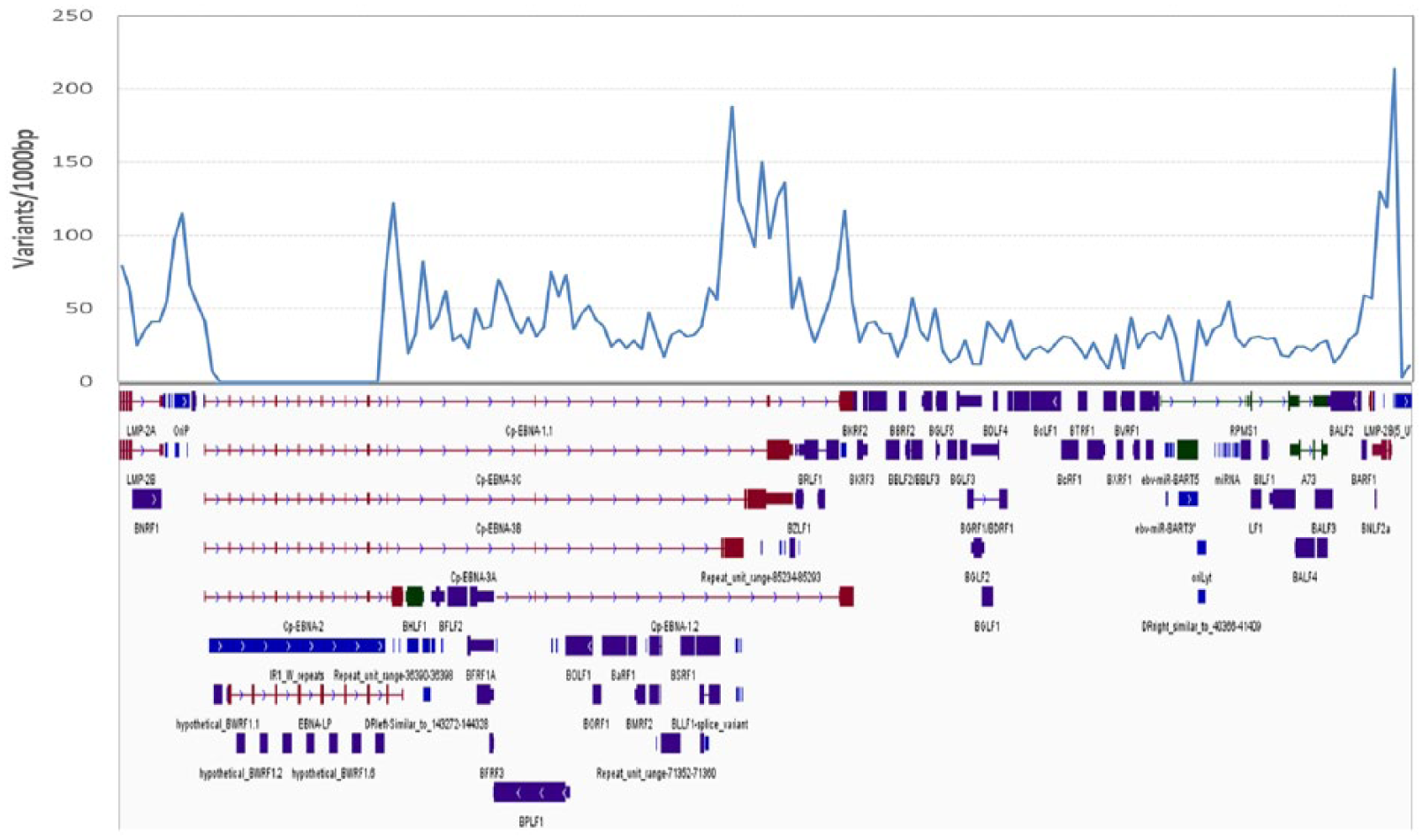
Regions encoding latent proteins have highest diversity across EBV genomes. Variant frequency across EBV genomes derived from 95 samples. The line graph is plotted across the genome showing the total number of variants in a sliding 1000-nt window.

Number of non-synonymous variants contained in the nine categories of EBV-encoded proteins. The majority of the amino acid changes are located in latency proteins and tegument proteins in all EBV strains.
Population structure of EBV strains
To identify major features of variation associated with geographic origin and oncogenic EBV subtypes, we performed Identity by State (IBS) analysis 20 on variants of 41 tumor samples and 54 healthy samples. The results showed that the strains extracted from gastric carcinoma patients with a geographic origin of South China (red), gastric carcinoma patients with a geographic origin of North China (black), and healthy donors with a geographic origin of South China (blue) tended to cluster together (Figure 3). A principal component analysis (PCA) was also performed on variations in the genomes. Principal component 1 (PC1) and 2 (PC2) were the greatest contributors (19.0% and 16.1% of the variance, respectively) to discriminate between the strains, and the top 10 PCs explained approximately 60% of the variance (Figure S1).

Cluster analysis on the 95 × 95 matrix of identity-by-state pairwise sequence distances of EBV whole genome. 95 EBV genomes are isolated from 41 gastric cancer patients in which 24 are from North China and 17 from South China and 54 healthy individuals in which 6 are from North China and 48 from South China.
Weighted correlation network analysis 21 was used to discover the candidate traits of samples that contributed to the clustering of strains. Clustering dendrograms of samples based on their Euclidean distance (Figure 4) demonstrated that there was a population structure. Northern strains and Southern strains clustered to different branches. In particular, gastric carcinoma strains and healthy control strains had very notable stratification, implying that certain EBV subtypes are associated with different cancer risks. For gastric carcinoma–isolated EBV (GC-EBV), in addition to the geographic origin, the other factors of sex, age, primary site, and differentiation status of tumor, tumor size, and virus load were not related to the clustering (Figure S2). The few samples that were not strictly clustered together according to their geographic origins or disease status may be attributed to viral recombination during the population migration.

Clustering dendrogram of 95 EBV genomes based on their Euclidean distance.
EBV variants and genes associated with gastric carcinoma
The oncogenic genes of EBV in the progression and development of gastric carcinoma have not been identified or well characterized. We conducted a genome-wide association study of 41 cases of GC-EBV and 54 controls of EBV isolated from healthy donors to distinguish the genes associated with gastric carcinoma by a logistic regression model adjusted for a mixture of each site, sex, age, and geographic origin. The hot association regions were EBNA3s, BBLF2/BBLF3, BPLF1, and BRLF1, which harbored 23/31, 3/12, 2/12, and 3/12 associated non-synonymous variants/variants, respectively (Figure 5(a), Table S5). The results indicated the very important role EBNA3s might play in gastric cancer development.

EBV whole genome association analysis. Manhattan plot of the genome-wide P values of association study. (a) Association was assessed by logistic regression analysis with adjustment for sex, age, and geographic origin. The −log10-transformed p values (y-axis) of 3684 SNPs and INDELs in 41 gastric carcinoma cases and 54 healthy controls are presented based on their positions on EBV genome. The smallest p value is 2.02 × 10−5. (b) Association was assessed by logistic regression analysis with adjustment for sex, age, and disease status. The −log10-transformed p values (y-axis) of 3684 SNPs and INDELs in 65 South China individuals and 30 North China individuals are presented based on their positions on EBV genome. The smallest p value is 2.46 × 10−5.
The EBV genes involved in the occurrence and development of gastric carcinoma and the oncogenic genes may serve as potential therapeutic targets during prevention, treatment, and vaccine development. We also identified genes using the SNP-set (sequence) Kernel association test (SKAT) analysis, 22 which aggregates individual SNP score statistics in a SNP set and efficiently computes SNP-set level p values with adjustments for sex, age, and geographic origin. BRLF1 (transcription activator23,24), BBRF3 (glycoprotein M, involved in envelope–tegument interaction25,26), and BBLF2/BBLF3 (primase-associated factor 27 ) had the most significant associations with gastric carcinoma (Table 2).
SKAT analysis of geneset of variants between gastric carcinomas and healthy controls.

SKAT: SNP-set (sequence) Kernel association test; N.Marker.All: number of variants of the gene; N.Marker.Test: number of calculated variants.
Identification of geographically associated EBV variants and genes
The geographic variants and relevant genes of EBV have not been well elucidated. Therefore, we combined GC-EBV isolates and healthy controls, of which 30 samples had North China origins and 65 samples had South China origins. We then conducted a viral genotype–phenotype association analysis using a logistic regression model that was adjusted for a mixture of each site, sex, age, and disease status. The viral genome-wide association study revealed that the top association sites were located in the BamHI fragment A rightward transcript (BART) microRNA (miRNA) region, the EBNA2 gene, and the EBNA1 gene, which harbored 75, 21, and 19 associated variants, respectively. Additionally, 14 and 13 non-synonymous variants were found in EBNA2 and EBNA1, respectively (Figure 5(b), Table S6). SKAT analysis was performed to test for associations with geographic origin of genes in the EBV genome, adjusted for sex, age, and disease status. The results showed that LMP1 (LMP, interferes with signaling, TRAF-binding through CTAR1 and 2, which is essential for immortalization 28 ) and BNLF2a (potential membrane protein 29 ) had the highest associations (Table 3), thus demonstrating the most significant roles these genes played in the evolution and distribution of EBV in populations of North and South China.
SKAT analysis of geneset of variants between South China individuals and North China individuals.

SKAT: SNP-set (sequence) Kernel association test; N.Marker.All: number of variants of the gene; N.Marker.Test: number of calculated variants.
Discussion
Genome-wide analysis of EBV sequence is essential for the understanding of viral infection, latency, and lytic replication and its association with diseases. In this study, we adopted the optimized virus-targeted capture and next-generation sequencing, successfully sequencing 41 EBV isolates from gastric carcinoma biopsies and 54 isolates from saliva of healthy donors. Here, we found that the latency-related genes were the most diverse regions of the viral genome and that the EBNA3A, BPLF1, EBNA3C, EBNA3B, and LMP1 genes harbored abundant non-synonymous variants, suggesting positive selection for these highly polymorphic genes. 12
We completed the virus genome-wide association study of EBVs from gastric cancers and healthy controls. Accordingly, EBNA3s, BBLF2/BBLF3, BPLF1, and BRLF1 genes were of interest, and they all had more than 10 associated variants. In particular, EBNA3s, including EBNA3A, EBNA3B, and EBNA3C, harbored 23 non-synonymous variants in all 31 associated variants, suggesting that the positive selection for these genes is associated with crucial impacts on tumorigenesis of EBVaGC. However, compared to other EBV-encoded nuclear antigens (EBNA1, EBNA2, and EBNA-LP), the roles of EBNA3s proteins in EBV biology are poorly understood. EBNA3s are essential for the latency of EBV in B-cells and participate in reprogramming the host genes to facilitate cell proliferation and differentiation as well as immune surveillance because EBNA3s are targets of immune recognition during the latent infection period. EBNA3A and EBNA3C are considered oncoproteins because they target tumor suppressor pathways in B-cell proliferation. However, EBNA3B is described as a tumor suppressor gene in EBV. 30 In regard to gene-level analysis, BRLF1, BBRF3, and BBLF2/BBLF3 were associated meaningfully with gastric carcinoma. BRLF1, which was also found to be a hot spot in an association analysis, encodes Rta to activate the expression of early genes that are essential for viral genome replication and induction of the lytic cycle. In addition to the latent cycle, the EBV lytic cycle contributes to carcinogenesis by inducing oncogenic cytokines and growth factors.31,32
We also identified the hot association spots and significant genes of EBV to explain the EBV population structure. The most association variants were located in the BART miRNA region, the EBNA2 gene, and the EBNA1 gene. BART miRNAs potentiate tumor growth in vivo. 33 Many were highly expressed in nasopharyngeal carcinoma and gastric cancer, contributing to viral latency, host cell survival, and immune escape.34–39 The associations of BART variation and geography are not comprehensively understood. EBNA1 is expressed consistently in all EBV-positive cancers and contributes to the maintenance of the EBV genome. 40 Based on the sequence variation of the C-terminus region of EBNA1, two main strains were identified. The EBNA1 of the prototype strain was similar to that of B95-8 with 3 amino acid (AA) substitutions; the variant strain differed from B95-8 by 15 AA substitutions. Moreover, based on the AA present at codon 487 of EBNA1, several subtypes were additionally characterized. Importantly, previous research showed that subtype V-val is dominant in Asia, in both EBV-associated malignancies and control populations, which suggests that it is a geographical polymorphism. 41 EBNA2 plays important roles in B-cell transformation and helps classify type 1 (EBV isolated from B95-8) and type 2 (EBV isolated from AG876), where type 1 has a longer reading frame. 42 Furthermore, EBV type 1 is detected more frequently in Asian and Caucasian populations,43,44 while type 2 is more prevalent in Africa. 45 SKAT analysis implied the importance of geographically associated genomic variations in LMP1 and BNLF2a genes. The LMP1 gene has three main polymorphisms, including the 30 bp deletion in the C-terminus, the XhoI loss in the N-terminus, and the AA changes in the C-terminus. These variations are more common in China than in other regions in the world. 46
Taken together, we described the global view of EBV genomic variations and determined the genetic variants and genes associated with gastric carcinoma and geography of North and South China. Our investigations provide insights into the genetic basis of EBV-associated gastric carcinogenesis and vaccine design for further studies.
Materials and methods
Study subjects
The tissue samples from tumor biopsies were collected from Sun Yat-sen University Cancer Center in Guangdong Province and The Affiliated Hospital of Qingdao University in Shandong Province from 2008 to 2014. The saliva samples were collected from healthy donors in Guangdong Province and Beijing City from 2010 to 2013. Sample details are listed in Table S1. DNAs were extracted using the DNeasy blood and tissue kit (QIAGEN, Hilden, Germany) according to the manufacturer’s instructions. All participants recruited in this study were informed and this study was approved by the institutional ethics committee of the Sun Yat-sen University Cancer Center.
Virus load
Real-time polymerase chain reaction (PCR) of a 202-bp amplicon of BALF5 was used to measure the virus load (forward primer, 5′-GGTCACAATCTCCACGCTGA-3′; reverse primer, 5′-CAACGAGGCTGACCTGATCC-3′). Samples with threshold cycle (Ct) values under 30 were sequenced.
Targeted capture and sequencing
First, 2–3 µg of genomic DNA was sheared to 150–200 bp fragments by Covaris S2 (Covaris, Woburn, MA, USA). Then, the fragments were end-blunted, “A”-tailed, and adaptor-ligated according to the manufacturer’s instructions (Illumina Inc., San Diego, CA, USA). The fragments underwent 9 cycles of PCR and product purification, and the libraries were hybridized with EBV capture probes designed by MyGenostics (Beijing, China) for 24 h at 65°C. The uncaptured fragments were removed by washing, and eluted targeted fragments were enriched by 18 cycles of PCR to generate libraries for sequencing. Libraries were quantified and sequenced for paired-ends using an Illumina HiSeq 2000 sequencer (Illumina Inc.).
Sequence alignment and variant calling
We first filtered low-quality reads and trimmed 3′/5′ adaptors using Trim Galore (quality score > 20, read length > 60 bp). Then, the clean reads were aligned to the reference genome EBV-WT (NC007605.1) using the Burrow Wheeler Aligner (BWA). 47 Aligned reads were converted to binary alignment map (BAM) format using SAMtools. 48 The reads were sorted, and the duplicates were removed using Picard Tools. The details of the sequencing qualities are summarized in Table S1. For variant calling, a Genome Analysis Toolkit (GATK) was used to improve alignments, call genotypes, and filter variants with optimized parameters. 49 BAM files were realigned by the GATK IndelRealigner module. The base quality of the mapped reads was recalibrated by the GATK BQSR module. Raw variants were called by the GATK UnifiedGenotyper module (with parameters stand_emit_conf 10.0, dcov 1000). Variants were filtered by the GATK VariantFiltration module (with parameters MQ0 >= 4 && ((MQ0/(1.0 * DP)) > 0.1) QUAL < 50.0 QD < 2.0 MQ < 40.0 FS > 250.0 for SNPs; MQ0 >= 4 && ((MQ0/(1.0 * DP)) > 0.1 QUAL < 50.0 FS > 200.0 for INDELs). Variant evaluation was made using the GATK VariantEval module. Variant annotation was performed by SnpEff 50 based on the annotation of NC007605.1 (NCBI annotation, November 2013). Variants include substitutions, insertions, and deletions.
IBS analysis and PCA
The analyses were performed on variants using the R package “SNPRelate.” 20 Variants with call rates less than 90% and located in repeat regions were disregarded.
EBV genome-wide association study
A logistic regression model was used to test the association between EBV variants and sample phenotype. Mixtures of each site, sex, age, and geographic origin (or disease status) were used as categorical covariates. Only variants with call rates >90% were recruited into the association study.
Biostatistics analyses
Weighted correlation network analysis was performed by the R package “WGCNA.” 21 The SKAT analysis was performed by the R package “SKAT.” 22 Variants with call rates >90% were included into the analyses.
Footnotes
Acknowledgements
We thank all patients, healthy donors, doctors, and nurses participating in this study. Y.Y., M.X. and L.L. contributed equally to this work.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the Key Program of the National Natural Science Foundation of China (No. 81430059); the National Basic Research Program of China (No. 2013CB910300); the Health & Medical Collaborative Innovation Project of Guangzhou City, China (No. 201400000001); the Foundation of the Ministry of Science and Technology of Guangdong Province (No. 2015B050501005); and the International Program for PhD Candidates, Sun Yat-sen University.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.