
Abstract
Despite great scientific advances have been achieved in cancer treatment in recent years, the death rate of bladder cancer has been staying at a high level. Metformin, a widely-used and low-cost diabetes medicine, might have the potential of anticancer. The aim of this study was to evaluate the effects of metformin on bladder cancer cells and to identify potential molecular targets and signaling pathways. Bladder cancer 5637 cells transfected with either pcDNA/UCA1 vector or pcDNA3.1 empty vector were treated with various doses of metformin for different periods of time, and then cell proliferation and glycolysis were assessed. Reverse transcription polymerase chain reaction and Western blotting were applied to examine the expression of long non-coding RNA UCA1 and mammalian target of rapamycin–signal transducer and activator of transcription pathway molecules. We found metformin inhibited bladder cancer cell proliferation in a dose- and time-dependent manner. UCA1-overexpressed 5637 cells showed increased proliferation and glycolysis compared with control cells. Metformin downregulated both endogenous and exogenous UCA1 expression, leading to the inhibition of mammalian target of rapamycin–signal transducer and activator of transcription 3–hexokinase 2 signaling pathway. Our study provided the first evidence that metformin inhibited proliferation and glycolysis in cancer cells through regulation of long non-coding RNA UCA1. The discovery also suggested the important roles of long non-coding RNA in chemoprevention, which is a property of metformin.
Introduction
Urothelial carcinoma of the bladder is one of the deadliest and costly diseases worldwide. It is ranked the fourth among the most common cancers in men in the United States. Despite the great scientific advances have been achieved in cancer treatment in recent years, the death rate of bladder cancer has been stable. 1 At diagnosis, 70%–80% of bladder cancer is non-muscle invasive and the rest is muscle-invasive. The treatment for bladder cancer is essentially surgical therapy, either transurethral resection or radical cystectomy, followed by multiagent cisplatin-based chemotherapy or immunotherapy. Some of the chemotherapeutic drugs have been in use for more than decades and show reduced effectiveness and severe side effects. To overcome this situation, there are several attempts to find more effective novel drugs to be used alone or in combination with the drugs already in use. Metformin is the first-line medication for treatment of type 2 diabetes. Encouraging results emerged from studies in the past 10 years indicate that metformin can potentially be used as an effective anticancer drug in various neoplasms,2–4 first reported in 2006. 5 In addition, metformin has been shown to increase the sensitivity of tumor cells to chemotherapeutic drugs, 6 synergizes the effect of the chemotherapeutic drugs while decreases toxicity, therefore prolongs remission of cancers. 7
Clinical studies on the association between diabetes mellitus (DM) and metformin use with prognosis and outcomes of bladder cancer found that patients with DM and bladder cancer, who did not take metformin, were at an increased risk of cancer recurrence and progression.8–10 However, results are still controversial. The usage of metformin was not associated with a decreased incidence of bladder cancer in two large cohort studies.11,12 The meta-analysis also suggested that although metformin might be associated with a significant reduction in the risk of cancer and cancer-related mortality in all malignancies, no association was observed in bladder cancer. 13 Furthermore, the underlying molecular mechanism of antineoplastic activity of metformin is still largely unknown.
Urothelial cancer associated 1 (UCA1), a long non-coding RNA (lncRNA), was originally identified in bladder carcinoma.14,15 Accumulating evidences indicate that the expression of various IncRNAs, including GHET116,17 and n336928, 18 are upregulated in bladder cancer. LncRNAs play important regulatory roles in tumorigenesis by promoting cell proliferation, colony formation, migration, and invasion.15,19 Therefore, our study aims to investigate the effects of metformin on the biological functions of bladder cancer cells and to evaluate whether they are mediated by regulation of UCA1, which in turn, affects mammalian target of rapamycin (mTOR) signaling pathway.
Materials and methods
Cell proliferation assay
In vitro growth rate of bladder cancer cells 5637 and UMUC2 was measured by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay. The MTT assay is widely used for measuring cell viability and drug cytotoxicity: It is based on the ability of viable cells to reduce MTT from a yellow water-soluble dye to a purple-insoluble formazan product. 20 Briefly, exponential-phase cells were digested with trypsin (Invitrogen, USA) and 200 µL of cell suspension was seeded on 96-well tissue culture plates at a density of 8000 cells per well in a 5% CO2 humidified incubator at 37°C. After 24 h, culture media were removed and 100 µL fresh media with various concentrations of metformin were added. At each specific time point, 10 µL of MTT reagent (Promega, USA) was added into each well. During a 4-h incubation, purple formazan product was formed, and then 150 µL of dimethyl sulfoxide (DMSO) was added and mixed to allow complete solubilization. Absorbance at 570 nm was detected and recorded using a Universal Microplate Reader (Bio-Tek Instruments, USA). In total, three to five wells were analyzed for each condition and the results were presented as the mean ± standard deviation (SD).
Clonogenic assay
Clonogenic assays were performed as described previously21–24 with slight modifications. Briefly, 5637 cells treated under different conditions were plated in triplicates at a density of 200 cells/well in a six-well plate and cultured at 37°C for 2 weeks. Clones were stained with Giemsa solution, and colonies with more than 50 cells were counted manually using an inverted microscope.
Semi-quantification of UCA1 RNA by reverse transcription polymerase chain reaction
Total RNA was extracted from 5637 cells using TRIzol reagent (Invitrogen) according to the manufacturer’s instructions, and 500 ng of total RNA was reverse transcribed with random hexamers using ImProm II reverse transcriptase (Promega). The resulting complementary DNA (cDNA) product was amplified using gene-specific primers and hot-start polymerase chain reaction (PCR) for 25 cycles. The primers for UCA1 were as follows—5′-CTCTCCATTGGGTTCACCATTC-3′ (forward) and 5′-GCGGCAGGTCTTAAGAGATGAG-3′ (reverse). The primers for 18S were as follows—5′-CAGCCACCCGAGATTGAGCA-3′ (forward) and 5′-GTAGCGACGGGCGGTGTG-3′ (reverse). 18S was used as a housekeeping gene. Three independent experiments were repeated.
UCA1 transfection
Full-length human UCA1 cDNA was cloned into pcDNA3.1 (Invitrogen) and digested with restriction enzymes BamHI and EcoRI (New England BioLabs, USA). We used the primers 5′-GAATTCTGACATTCTTCTGGACA ATGAGTC-3′ (EcoRI) and 5′-GGATCCGGCATATTAGCT TTAATGTAGGTG-3′ (BamHI) to obtain full-length UCA1 cDNA from 5637 cells. 25 The recombinant plasmid construct pcDNA/UCA1 was then transfected into 5637 cells using FuGENE HD (Roche, Germany), and empty pcDNA3.1–transfected cells were used as a negative control (MOCK). The positive clone was identified by reverse transcription PCR (RT-PCR) of UCA1 and neo gene expression, and the fidelity of the UCA1-cloned sequence was confirmed with DNA sequencing. The primers for neo were as follows—5′-ACAAGATGGATTGCACGCAGG-3′ (forward) and 5′-TTCTCGGCA-3′ (reverse).
Western blotting
Western blotting was carried out using the protocol described previously with minor modifications.26–28 Cells were pelleted and then lysed by radioimmunoprecipitation assay (RIPA) buffer containing protease and phosphatase inhibitors (Roche, Switzerland). A volume of 40 µg of protein was subjected to electrophoresis with a 12% sodium dodecyl sulfate (SDS)-polyacrylamide gel (Bio-Rad, USA) and then transferred to a polyvinylidene difluoride (PVDF) membrane. Membranes were briefly washed with Tris-buffered saline (TBS) and blocked with 5% skim milk in TBS containing 0.05% Tween 20 (TBST) for 1 h at room temperature, followed by incubation for overnight at 4°C with the corresponding antibodies in blocking bluffer: mTOR (Pierce, USA), phosphorylated mTOR (p-mTOR; Pierce), signal transducer and activator of transcription 3 (STAT3; Cell Signaling Technology, USA), p-STAT3 (Cell Signaling Technology), hexokinase 2 (HK2; Cell Signaling Technology), p-HK2 (Cell Signaling Technology), and β-actin (Cell Signaling Technology). Membranes were then washed three times with TBST and probed with appropriate horseradish peroxidase–conjugated secondary antibody (Santa Cruz Biotechnology, USA) for 1 h. The antigen–antibody reaction was detected by enhanced chemiluminescence substrate and analyzed by Quantity One Discovery software (Bio-Rad) according to the manufacturer’s instructions. Data presented were representative of three separate experiments.
Lactate concentration measurement
The Bladder cancer 5637 cells treated with various concentrations of metformin were incubated in complete media with 10% fetal bovine serum (FBS) at 37°C under 5% CO2. After 48 h of incubation, the cells were disrupted and centrifuged. Supernatant was recovered and used to measure lactate concentration using Lactate Detection Kit (Apply GEN, China) according to the manufacturer’s protocol. Lactate production was calculated based on the standard curve and normalized to the cell number.
Small interfering RNA knockdown experiments
Both the UCA1 small interfering RNA (siRNA) and the scrambled control siRNA were synthesized by Invitrogen. The sequence of si-UCA1 was 5′-GTTAATCCAGGAGACAAAGA-3′. 29 The experiment was carried out using the protocol described previously.28,30,31
Statistical analysis
Data were showed as mean ± SD. Statistical comparisons were analyzed by Student’s t test using SPSS 19.0 software (SPSS Inc., USA). p < 0.05 was considered as statistically significant.
Results
Metformin inhibits proliferation of bladder cancer cells
Previous study showed that metformin inhibited proliferation of bladder cancer cells via activation of adenosine monophosphate (AMP)-activated protein kinase (AMPK) and suppression of mTOR. 32 To verify this effect in our system, various concentrations of metformin (5–20 mM) were added to bladder cancer cells 5637 and UMUC2 for up to 72 h. Dose-response curve (Figure 1(a) and Supplementary Figure 1) indicated that metformin inhibited 5637 cell proliferation in a dose-dependent manner. Significant inhibition (p < 0.05) was observed starting at 10 mM. The time course (Figure 1(b)) showed promising inhibitory effects for all the doses, except 5 mM, after 48 h of treatment. These findings confirmed that metformin inhibited proliferation of bladder cancer cells.

Effects of metformin on the proliferation of bladder cancer cells. (a) Dose-response analysis of the effect of metformin on the proliferation of 5637 cells. Exponential-phase 5637 cells maintained in 10% fetal bovine serum were exposed to the indicated concentrations of metformin for up to 72 h, and then the cell proliferation was measured. (b) Time-course analysis of the effect of metformin on the proliferation of 5637 cells. The cells were treated with 5–20 mM of metformin, and cell proliferation measurements were performed at the indicated times (n = 3–5, *p < 0.05).
Metformin decreases UCA1 expression in bladder cancer cells
UCA1 was upregulated in bladder tumors while there was no expression in normal bladder tissues. The proliferation, invasion, and drug resistance behaviors of bladder cancer cell line were enhanced by exogenous UCA1 expression. 15 Therefore, we hypothesized that the inhibition of cell proliferation by metformin may be mediated by the downregulation of UCA1. To test this hypothesis, we detected the UCA1 RNA level under the treatment of metformin. As shown in Figure 2, the expression of UCA1 was significantly inhibited by metformin at 10 mM (p < 0.05), and it was further inhibited by 20 mM of metformin (p < 0.05). These results indicated that the antineoplastic activity of metformin might be associated with its regulation on UCA1.

Reduction of UCA1 expression by metformin in bladder cancer cells. (a) UCA1 RNA levels from 5637 cells treated with vehicle, 10 mM, and 20 mM of metformin were determined by RT-PCR. A representative agarose gel electrophoresis of PCR products for the detection of UCA1 (upper panel). 18S RNAs, which were included to serve as internal controls, were shown in the lower panel. (b) Quantitative results of UCA1 RNA from three independent experiments (*p < 0.05, compared with vehicle group).
Metformin inhibits proliferation of UCA1-transfected bladder cancer cells
To further investigate whether the antineoplastic activity of metformin mediated the modulation of UCA1, bladder cancer 5637 cells were transfected with pcDNA/UCA1 and pcDNA empty vector (MOCK) as negative control. We first confirmed that UCA1 expression was significantly increased in pcDNA/UCA1-transfected 5637 cells in contrast to MOCK cells (Figure 3(a)). Compared with empty vector–transfected cells, the pcDNA/UCA1-transfected cells had an increased proliferation rate as measured by MTT assay (Figure 3(b)). The cell viabilities at 48 and 72 h in UCA1-overexpressed cells were significantly higher than those in MOCK cells (Figure 3(b) left panel; n = 3, *p < 0.05). These results indicated that transfection with exogenous UCA1 resulted in overexpression of UCA1, which in turn, promoted cell proliferation in bladder cancer cells.
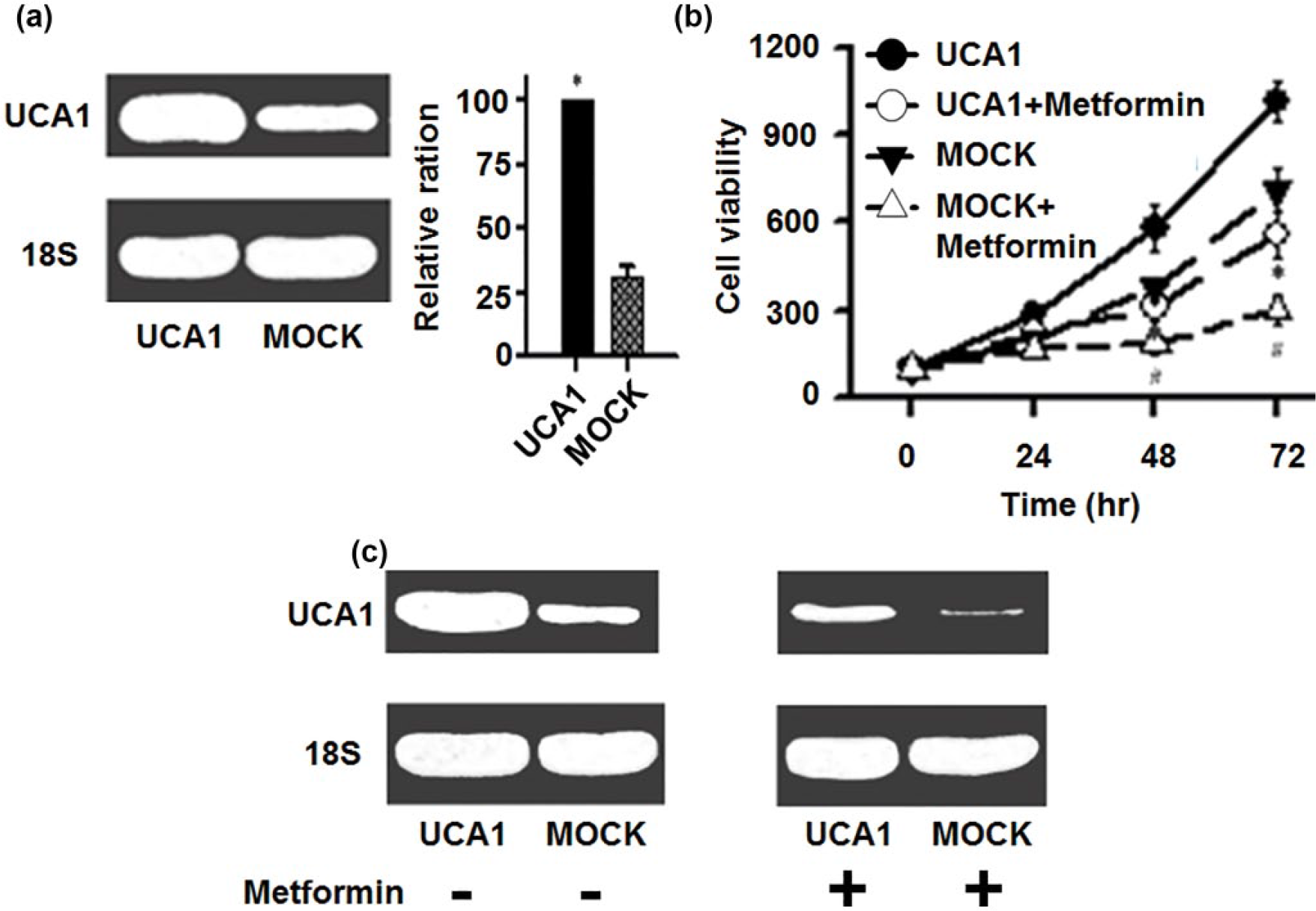
Effects of metformin on the proliferation of UCA1-overexpressed bladder cancer cells. (a) Total RNA was isolated and the level of UCA1 was determined using RT-PCR. A representative agarose gel electrophoresis image was shown on the left and the quantitative results of UCA1 RNA from three independent transfection experiments were shown on the right (*p < 0.05). (b) Cell proliferation in UCA1-overexpressed 5637 cells and MOCK cells treated with or without metformin as measured by MTT assay (left panel). Compared with empty vector–transfected cells, the UCA1-transfected cells had increased proliferation rate. Metformin treatment for 48 and 72 h significantly decreased cell proliferation in both the UCA1-overexpressed bladder cancer cells and MOCK cells (n = 3, *p < 0.05). (c) A representative agarose gel electrophoresis image of UCA1 expression in UCA1-overexpressed 5637 cells and MOCK cells treated with or without metformin is shown in right panel.
Next, we examined the effect of metformin on the tumorigenic behavior of UCA1-transfected cells. Similar to what we observed in the non-transfected 5637 cells, the expression levels of UCA1 in both pcDNA/UCA1-transfected cells and MOCK cells were profoundly suppressed by metformin (Figure 3(b), right panel). Accordingly, the proliferation rates of both pcDNA/UCA1 and MOCK cells treated with metformin were significantly lower than those of the corresponding untreated cells (Figure 3(b), left panel; n = 3, *p < 0.05). The results demonstrated that metformin is sufficient to downregulate both the endogenous and exogenous UCA1 expression, which leads to suppression of proliferation in bladder cancer cells.
Modulation of mTOR pathway by metformin in UCA1-transfected cells
mTOR is a ubiquitous serine/threonine kinase that plays pivotal roles in mammalian metabolism and physiological processes including cell survival, growth, proliferation, transcription, and autophagy. 33 Recent study finds that UCA1 plays a positive role in cancer cell glucose metabolism by activating mTOR signaling pathway. 34 Therefore, we predicted that metformin might modulate mTOR signaling pathway via UCA1. To validate this hypothesis, UCA1-overexpressed 5637 cells and MOCK cells were treated with 15 mM metformin for 0–72 h. Protein levels of p-mTOR, mTOR, and two mTOR pathway downstream targets (p-STAT3 and p-HK2) were analyzed by Western blotting. Our data clearly showed that activated p-mTOR was inhibited by metformin after 48 h of treatment in UCA1-overexpressed bladder cancer cells, while no change was observed for total mTOR level (Figure 4(a), upper left panel). mTOR downstream target p-STAT3 was also inhibited by metformin. Interestingly, this effect appeared at an earlier time point (24 h). Hexokinases catalyze the first and rate-limiting step of glycolysis, and hexokinase 2 (HK2), a key mediator of aerobic glycolysis and promoter of tumor growth, is expressed in insulin-sensitive tissues and overexpressed in many tumor cells including bladder cancer. 35 The relative ratio of p-HK2/HK2 started to decline after 24 h of metformin treatment and decreased to about 10% at 72 h (Figure 4(b), lower left panel), indicating that the mTOR signaling pathway was inhibited by metformin. Stronger inhibitory effects were also observed in empty pcDNA3.1–transfected cells (Figure 4(a) and (b), upper right panel and lower right panel), further confirming that metformin modulated the mTOR signaling pathway in bladder cancer cells by downregulating both endogenous and exogenous UCA1 expression.

Modulation of mTOR pathway by metformin in UCA1-transfected bladder cancer cells. (a) UCA1-overexpressed 5637 cells and MOCK cells were treated with 15 mM of metformin for up to 72 h. Cellular proteins were extracted at indicated time points. Both total and phosphorylated forms of mTOR signaling pathway proteins were detected by immunoblotting analysis using the corresponding antibodies. A representative blot of three independent experiments was shown. (b) The densities for Western blot bands were quantified by Quantity One Discovery software (Bio-Rad, USA). The ratios of density for p-mTOR/mTOR, p-STAT3/STAT3, and p-HK2/HK2 were calculated for each lane, and the value for time 0 was considered as 100% and compared with other time points for each experiment. Data were expressed as mean ± SD (n = 3, *p < 0.05).
Metformin suppresses glycolysis in bladder cancer cells
Given the profound inhibitory effect of metformin on HK2, the rate-limiting enzyme of glycolysis, we investigated the changes in glycolysis in bladder cancer cells treated with metformin. The 5637 cells were treated with 15 mM metformin for 48 h. Cells were disrupted and the lactate concentration was measured. As shown in Figure 5(a), metformin significantly reduced the lactate production in MOCK cells (n = 3, p < 0.05). Similar suppression effect was also observed in UCA1-overexpressed cells, in which metformin treatment reduced the lactate level to about 20% of that in untreated cells (p < 0.05). Interestingly, the lactate levels in pcDNA/UCA1-transfected 5637 cells before and after metformin treatment were both much higher than those in MOCK cells (p < 0.05), and the similar results were also found in cells with si-UCA1 (Supplementary Figure 2 and Figure 5(b)), reflecting a relatively activated mTOR signaling pathway and upregulation of p-HK2 level as shown in Figure 4.

Metformin suppresses glycolysis in bladder cancer cells. UCA1-transfected 5637 cells and MOCK cells treated with or without 15 mM of metformin were incubated in complete media with 10% FBS at 37°C under 5% CO2. After 48 h of incubation, the cells were disrupted and centrifuged. Supernatant was recovered and used to measure lactate concentration using Lactate Detection Kit (CMA Microdialysis AB, USA) according to the manufacturer’s protocol. (a) The value for UCA1-overexpressed cell without metformin treatment was considered as 100% (n = 3, *p < 0.05, compared to UCA1-overexpressed cell; #p < 0.05, compared to MOCK cell). (b) The value for scramble cell without metformin treatment was considered as 100% (n = 3, *p < 0.05, compared to scramble cell; #p < 0.05, compared to si-UCA1 cell).
Discussion
Metformin is the most commonly prescribed antihyperglycemic agent for patients with type 2 DM due to its established treatment efficacy, good safety profile, and low cost. Several clinical studies have reported an association between metformin and reduced cancer risk and/or improved cancer survival.13,36,37 Laboratory research also provides converging evidence that metformin has antineoplastic activity. Early studies showed that metformin acts as a growth inhibitor rather than an insulin sensitizer for breast cancer cells.5,38 Administration of metformin significantly decreased the growth of pancreatic cancer cells in nude mice 39 and suppressed intestinal polyp growth in ApcMin/+ mice. 40 However, the effect of metformin on bladder cancer has just emerged recently and the results are controversial. While Goossens et al. 12 found that metformin had no influence on the risk of bladder cancer compared with sulfonylurea in type 2 DM patients, others showed an association between metformin use and improved recurrence-free survival and bladder cancer–specific survival in diabetic patients undergoing radical cystectomy.9,41 In this study, we showed that metformin inhibited the proliferation of bladder cancer cells in a time- and dose-dependent manner. Consistent with Zhang’s finding that metformin significantly inhibited the proliferation and colony formation of bladder cancer cells, 32 our observations provide, at least partially, the laboratory explanation for improved survival in bladder cancer patients treated with metformin.
Next, we investigated the underlying mechanism by which metformin inhibits the proliferation of bladder cancer cells. The anticarcinogenic effects of metformin are postulated to be associated with both direct and indirect effects of the drug. 42 The indirect mechanism is due to the reduction of the hyperglycemia and hyperinsulinemia characteristics of type II diabetes, leading to reduced insulin-receptor activation and proliferation of the subset of neoplasms for which hyperinsulinemia provides a growth advantage. 43 The direct effect of metformin is primarily mediated by inhibiting complex I of the mitochondrial respiratory chain, which results in impaired mitochondrial function and an increase in the ratio of AMP/adenosine triphosphate (ATP). Increased levels of AMP activate AMPK, leading to a reduction in mTOR signaling and relative protein synthesis in cancer cells.4,44 However, metformin may also present its beneficial effect on cancer prevention and treatment through other targets and further research into the exact mechanism of the action of metformin is ongoing. LncRNAs, defined as transcripts of with more than 200 nt with no protein coding function, have drawn growing attention in many fields in the last decade. Several studies have confirmed lncRNA UCA1 to be a biomarker for bladder cancer.45–47 Moreover, UCA1 is increasingly considered to play a pivotal role in the development and progression of urothelial carcinoma. Overexpression of UCA1 in bladder cancer cell line significantly enhances the tumorigenesis, invasion potential, and drug resistance, both in vitro and in vivo. 15 Furthermore, UCA1 regulates cell cycle progression through CREB (cyclic AMP response element binding) via phosphoinositide 3-kinase (PI3K)/AKT-dependent pathway in bladder cancer. 48 Two pieces of evidence support our hypothesis that metformin regulates the biological functions of bladder cancer cells through modulation of UCA1. First, UCA1 is overexpressed in urothelial tumor14,15 and has been shown to promote glycolysis by upregulating HK2 via mTOR–STAT3 pathway, which in turn contributes to the rapid growth of tumor cells. 34 Second, metformin activates AMPK and inhibits the activation of mTOR which leads to a reduced cellular proliferation in bladder cancer cells, 32 and the AMPK-mediated mTOR inhibition is considered as the crucial factor responsible for the antitumor properties of metformin. 49 Here, we demonstrated for the first time that metformin inhibited the expression of UCA1 in cancer cells. Furthermore, to achieve overexpression of UCA1, we transfected 5637 cells with pcDNA/UCA1. Interestingly, in these UCA1-overexpressed bladder cancer cells, metformin also inhibited UCA1 expression and reduced cell proliferation effectively.
As suggested by recent findings, UCA1 promotes cell proliferation by activating the PI3K-Akt-mTOR pathway 48 and increases glycolysis through mTOR–STAT3 pathway in bladder cancer. 34 Therefore, we speculated that metformin might modulate the function of 5637 cells through inhibition of UCA1-mTOR-STAT3 signaling pathway. As expected, we found that the activation and phosphorylation of mTOR and STAT3 were positively related to UCA1 levels in both UCA1-overexpressed cells and MOCK cells. Metformin treatment inhibited UCA1 expression in a time-dependent manner. Accordingly, the levels of p-mTOR and p-STAT3 were reduced, paralleled to the levels of UCA1. Since STAT3 is a direct transcriptional activator for HK2,34,50 we examined the activation of HK2, the major isozyme that is overexpressed in tumors and contributes to aerobic glycolysis, and the alteration of glycolysis in our system. As phosphorylation and activation of STAT3 were inhibited by the treatment of metformin via UCA1-mTOR pathway, the level of p-HK2 and the relative ratio of p-HK2/HK2 decreased significantly. Moreover, the lactate production, an indicator for glycolysis, was significantly reduced under the treatment of metformin in both UCA1-overexpressed 5637 cells and MOCK cells. Our results not only support that the UCA1-mTOR-STAT3 signaling pathway plays a pivotal role in the activation of HK2 and promoting glycolysis in cancer cells, but also reveal a novel mechanism underlying the antitumor action of metformin.
Taken together, our study here provided the first evidence that metformin inhibits aerobic glycolysis in cancer cells through the regulation of lncRNA UCA1, which in turn modulates mTOR-STA3-HK2 signaling pathway. Though additional in vivo evaluation is required, this novel linkage of metformin and lncRNA may encourage the findings of novel targets for the therapeutic strategies of bladder cancer.
Footnotes
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.