
Abstract
Angiogenesis plays important roles in progression of hepatocellular carcinoma. The antiangiogenic mechanisms of vitexicarpine are not fully defined. Therefore, we conducted the following study to evaluate the antiangiogenic mechanism and antitumor activity of vitexicarpine in vivo model of hepatocellular carcinoma through modulation of vascular endothelial growth factor signaling pathway. Hepatocellular carcinoma was induced in Sprague Dawley rats by thioacetamide. Hepatocellular carcinoma was assessed by measuring serum alpha-fetoprotein and investigating liver sections stained with hematoxylin/eosin. Hepatocellular carcinoma rats were injected with vitexicarpine (150 mg/kg) for 2 weeks. Hepatic vascular endothelial growth factor was measured by enzyme-linked immunosorbent assay. Protein and expression of hepatic phospho-Ser473-AKT (p-AKT) and phospho-Tyr419-Src (p-Src) were determined. The apoptotic pathway was evaluated by assessment of protein expression of caspase-3. Vitexicarpine increased rats’ survival time and decreased serum alpha-fetoprotein as well as it ameliorated fibrosis and massive hepatic tissue breakdown. It attenuated hepatocellular carcinoma–induced protein and gene expression of vascular endothelial growth factor, p-AKT, p-Src, and caspase-3. In conclusion, this study suggests that vitexicarpine possesses both antiangiogenic and antitumor activities through inhibition of vascular endothelial growth factor, p-AKT/AKT, and p-Src with subsequent inhibition of apoptotic pathway.
Introduction
Hepatocellular carcinoma (HCC) is a mutual, malignant tumor worldwide. Most cases of HCC develop in the background of liver cirrhosis. 1 Although sorafenib, vascular endothelial growth factor (VEGF) receptor and tyrosine kinase inhibitor, has been shown to extend the survival by nearly 3 months in patients with progressive HCC,2,3 its therapeutic potential is restricted due to severe side effects and extraordinary high cost.4,5 There are critical needs to explore possible alternative strategy that may effectively control HCC.
Angiogenesis is a key regulator of tumor growth and metastasis. 6 Tumor angiogenesis may be controlled by angiogenic factors such as VEGF pathway through AKT and Src especially in HCC.7,8 Various substances in Chinese medicinal herbs are assumed to be potent kinase inhibitors and demonstrated a preventive effect on cancer; 9 among these herbs is vitexicarpine which is a flavonoid extracted from the fruits of Vitex rotundifolia. 10 It has cytotoxic properties against human cancer cell lines 11 and inhibits the tumor necrosis factor-α (TNF-α)-induced vascular inflammation and T-lymphocyte proliferation. 12 It has been suggested that vitexicarpine could inhibit tumor growth and inflammation responses by negatively regulating angiogenesis. The antiangiogenic mechanism of vitexicarpine is not fully understood. Therefore, we conducted the following study to evaluate the vitexicarpine antiangiogenic activity and the extent of improvement of HCC in vivo model.
Materials and methods
Animals and their treatment outlines
The animal protocol was approved by the ethical committee of Faculty of Pharmacy, Mansoura University, Mansoura, Egypt. All experiment procedures were conducted in compliance with the National Institutes of Health’s Guide for the Care and Use of Laboratory Animals (NIH publication no. 8023, revised 1978).
Thirty-one adult Sprague Dawley males weighing 180–200 g were kept for a week for acclimatization under standard conditions of temperature (25°C–27°C), with a regular 12-h dark–light cycle and allowed free access to standard rat food. Rats were classified into below groups.
Control group
Ten rats were allowed free access to standard rat food for 16 weeks and then were given normal saline containing dimethyl sulfoxide (DMSO; dilution 7:1; Sigma–Aldrich Chemicals Co., St Louise, MO, USA) by intra-peritoneal (i.p.) injection every other day in the 17th and 18th weeks and served as a control group.
HCC group
Twenty-one rats were given 200 mg/kg thioacetamide (Sigma–Aldrich Chemicals Co.) in normal saline by i.p. injection twice a week for 16 weeks. 13
Vitexicarpine-HCC group
After week 16, eight rats of HCC group were chosen randomly and were given 150 mg/kg vitexicarpine in normal saline containing DMSO (7:1) by i.p. injection every other day for 2 weeks. 14
Collection of rats’ samples
At the end of the study period, blood samples were collected by retro-orbital method and centrifuged at 4000 r/min for 5 min to collect sera, which were stored at −80°C. Rats were killed by decapitation, and then livers were removed, weighed, and divided into five parts. The first part was immediately immersed in liquid nitrogen and stored at −80°C for assessment of gene expression of total AKT and Src. The second part was homogenized in 10 mM phosphate-buffered saline (PBS), pH 7.4, for determination of malondialdehyde (MDA) and superoxide dismutase (SOD). The third part was fixed in 10% buffered formalin for morphological and immunohistochemical studies. The fourth part was homogenized in lysis puffer (Abcam, Cambridge, UK), for the determination of VEGF by rat enzyme-linked immunosorbent assay (ELISA). The last part was homogenized in radio immuno precipitation assay (RIPA) buffer for estimation of the protein expression of phospho-Ser473-AKT (p-AKT), phospho-Tyr419-Src (p-Src), and caspase-3.
Morphological analysis
Liver pieces fixed in 10% buffered formalin were embedded in paraffin. Five-micrometer-thick sections were cut and stained with Mayer’s hematoxylin and eosin (H/E) for examination of cell structure by light microscope. Liver specimens were anonymously coded and examined in a blinded manner. The morphological changes were photographed using a digital camera–aided computer system (Nikon Digital Camera, Japan).
Immunohistochemistry
Immunohistochemical analyses were performed on 5-µm-thick sections cut from a paraffin block of liver. Sections were deparaffinized by heating and rehydrated using xylene and descending concentrations of ethanol. Antigen retrieval was performed in citrate buffer solution (pH 6.0). Endogenous peroxidase was blocked with 0.1% Triton for 15 min. All non-specific binding sites were blocked using 1% bovine serum albumin for 1 h at 22°C. Sections were incubated overnight with polyclonal p-AKT and p-Src (1/500 in blocking buffer; Abcam) for 16 h at 4°C. Then sections were incubated with an Alexa Fluor® 488–conjugated goat anti-rabbit polyclonal (1/1000; Thermo Fisher Scientific, Rockford, USA). The chromogen used was 2% 3,3′-diaminobenzidine (DAB) in 50 mM Tris buffer (pH 7.6). Slides were counter stained with H and examined under microscope (Nikon Digital Camera).
Assessment of liver function
Serum alanine aminotransferase (ALT), alkaline phosphatase (ALP), and gamma glutamyl transferase (GGT) activities as well as albumin concentration (Diamond Diagnostics, Giza, Egypt) were measured using standard colorimetric methodologies.
Assessment of oxidative stress
Oxidative stress was estimated in the liver homogenates through the following parameters:
MDA level was measured as described previously. 15 In brief, after precipitation of proteins by trichloroacetic acid, thiobarbituric acid reacts with MDA to form thiobarbituric acid–reactive substance that is measured at 532 nm.
SOD activity was determined using phenazine methosulfate method, 16 in which the ability of the enzyme to inhibit the phenazine methosulfate–mediated reaction of nitro blue tetrazolium dye is measured as an indication of the activity of SOD.
ELISA
The levels of biochemical parameters in liver homogenate and serum were measured by ELISA using a commercially available VEGF and alpha-fetoprotein (AFP), respectively (Abcam and USCN Life Science Inc., Houston, TX, USA, respectively) in accordance with the manufacturer’s instructions.
Quantitative, reverse transcription–polymerase chain reaction
Gene expression of total AKT, total Src, and caspase-3 in different rats’ hepatic tissues was determined using Maxima SYBR Green/Fluorescein qPCR Master Mix by Rotor-Gene Q (Qiagen, Hilden, Germany). Rat glyceraldehyde 3-phosphate dehydrogenase (GAPDH) was used as a housekeeping gene and an internal reference control. Gene-specific polymerase chain reaction (PCR) primers were designed using Primer Express 3.0 (Applied Biosystems, Foster City, CA, USA) according to the nucleotide sequence obtained from the Gene Bank. Thermal cycling conditions included initial activation step at 95°C for 10 min followed by 30–40 cycles of 94°C for 15 s, 55°C or 58°C for 30 s, and 72°C for 1 min. The primer sequences, amplicon size, and annealing temperatures used for amplification are listed in Table 1. Melting curve analysis of the PCR products was performed to verify their specificity and identity. Rotor-Gene Q (Qiagen) collected data automatically and analyzed the value of threshold cycle (Ct). Rat total AKT, total Src, caspase-3, and GAPDH messenger RNA (mRNA) relative expression were determined using 2−ΔΔCt method. PCR products were confirmed by running on 1.2% agarose gel electrophoresis.
The primer sets used for the detection of rat GAPDH, Src, and AKT.

GAPDH: glyceraldehyde 3-phosphate dehydrogenase; F: forward; R: reverse.
All primers are rat primers.
Western blot analysis
Liver tissue homogenates were prepared in ice-cold RIPA buffer from various studied rat groups. Protein concentrations of supernatants were determined by Bio-Rad protein assay (Hercules, CA, USA). Protein denaturation was achieved by heating at 100°C for 5 min in the presence of 2× Laemmli sample buffer (4% sodium dodecyl sulfate (SDS), 10% 2-mercaptoethanol, 20% glycerol, 0.004% bromophenol blue, and 0.125 M Tris–HCl). The samples were then subjected to SDS–polyacrylamide gel electrophoresis (PAGE). 17 The protein profiles were electroblotted onto the nitrocellulose membranes (Millipore Inc., Billerica, MA, USA) at 4°C overnight. The antibody incubation procedures and the blocking buffer used were as per the antibody supplier’s instructions (Abcam). Image was acquired using darkroom development techniques for normal image scanning methods for colorimetric detection. Various targets were normalized using β-actin as the loading control.
Statistical analysis
The mean ± standard error (SE) was used for descriptive statistics of quantitative variables. For comparison of means between groups, one-way analysis of variance (ANOVA) was used. Once the differences exist among the means, post hoc Bonferroni correction test was calculated. Kaplan–Meier method was used for calculation of rats’ survival. Statistical computations were done using the computer software SPSS version 13 (Chicago, IL, USA), Microsoft Excel 2010, and statistical significance was predefined as p <0.05.
Results
Vitexicarpine reduced HCC-induced VEGF expression
HCC caused 2.6-fold increase in VEGF protein expression as compared with control group. Treatment with vitexicarpine partially reversed this effect in HCC (Figure 1).
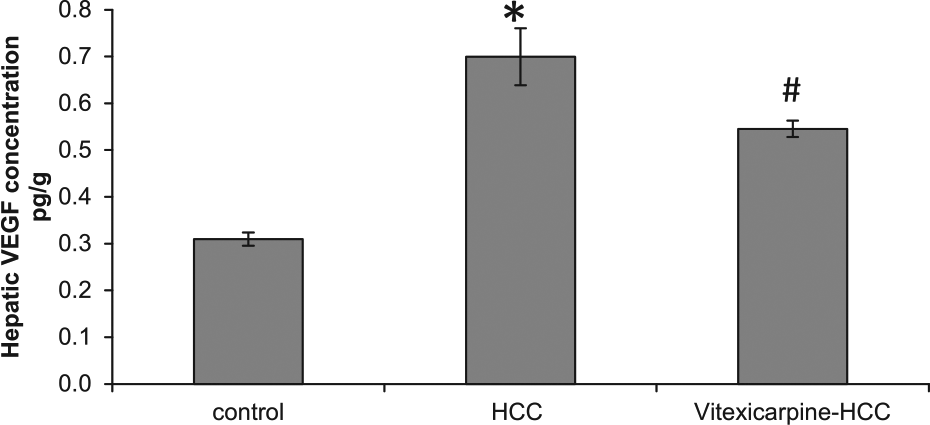
Vitexicarpine blocked vascular endothelial growth factor (VEGF) expression in vivo. Statistical analysis of VEGF concentration measured by ELISA showed a significant increase in HCC group (2.6-fold) compared with the control group. HCC rats treated with vitexicarpine blocked VEGF expression.
Antitumor activity of vitexicarpine
No death of rats was observed during treatment with vitexicarpine compared with more than 80% death in HCC group (Figure 2(a)). To confirm vitexicarpine’s antitumor activity, serum AFP level was measured in all animal groups (Figure 2(b)). Treatment with vitexicarpine significantly reduced HCC-induced elevation of AFP in HCC groups. There was also a notable increase in body mass and decrease in liver weight of vitexicarpine–HCC group (1.3- and 0.6-fold, respectively) as compared with HCC group.

Antitumor activity of vitexicarpine. (a) Kaplan–Meier survival curves of rats. (b) Treatment with vitexicarpine significantly reduced HCC-induced elevation in serum AFP in HCC group. *Significant difference as compared with control group at p < 0.05. #Significant difference as compared with HCC group at p < 0.05.
Amelioration of vitexicarpine on HCC-induced disturbance in liver function
HCC elevated the activities of both ALT and GGT (14.1- and 6.6-fold, respectively) associated with decreased albumin concentration (37%) as compared with control group (Figure 3). Vitexicarpine treatment significantly improved the liver function in HCC.

Effect of vitexicarpine on liver function. HCC rats treated with vitexicarpine showed a significant decrease in (a) serum alanine aminotransferase (ALT) and (b) gamma glutamyl transferase (GGT) as well as a significant increase in (c) serum albumin level as compared with HCC group.
Vitexicarpine ameliorates HCC-induced fibrosis
The improvement of the liver function by vitexicarpine was associated with hepatic morphological restoration. Histological examination of liver sections from HCC rats stained with H/E showed marked fibrosis and massive breakdown of hepatic tissues. However, sections from HCC rats treated with vitexicarpine showed nearly normal appearance of hepatic lobule (Figure 4).
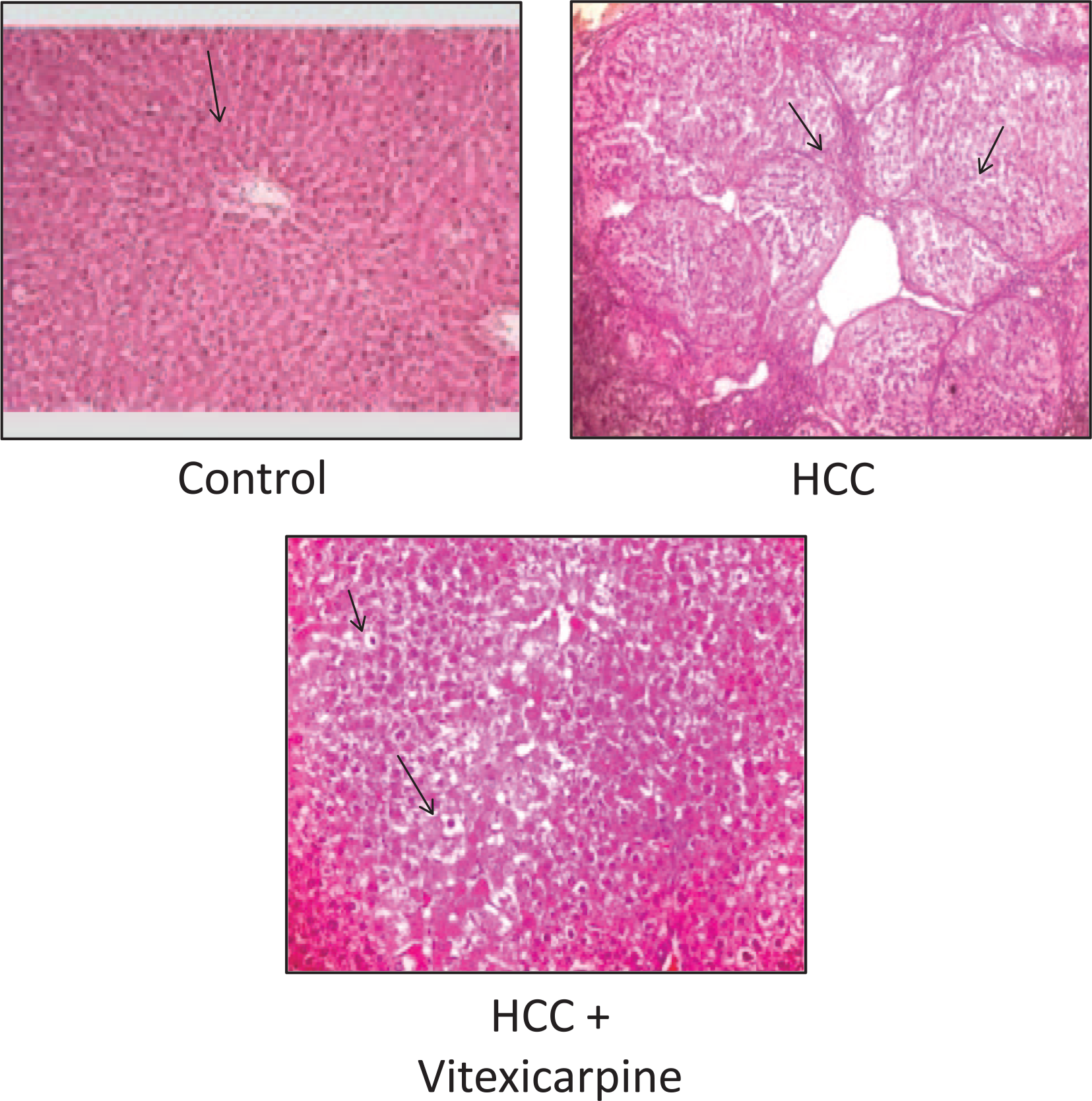
Hematoxylin and eosin–stained liver sections showed normal control, HCC, and vitexicarpine–HCC groups.
Vitexicarpine attenuated HCC-induced oxidative stress
The oxidative stress status has an important role in HCC development and progression. MDA and SOD measurements in liver homogenate samples showed a significant increase in MDA concentration (1.5-fold) and a significant decrease in SOD activity (27%) in HCC group as compared with the control group. Vitexicarpine treatment reversed these effects in HCC group (Figure 5).
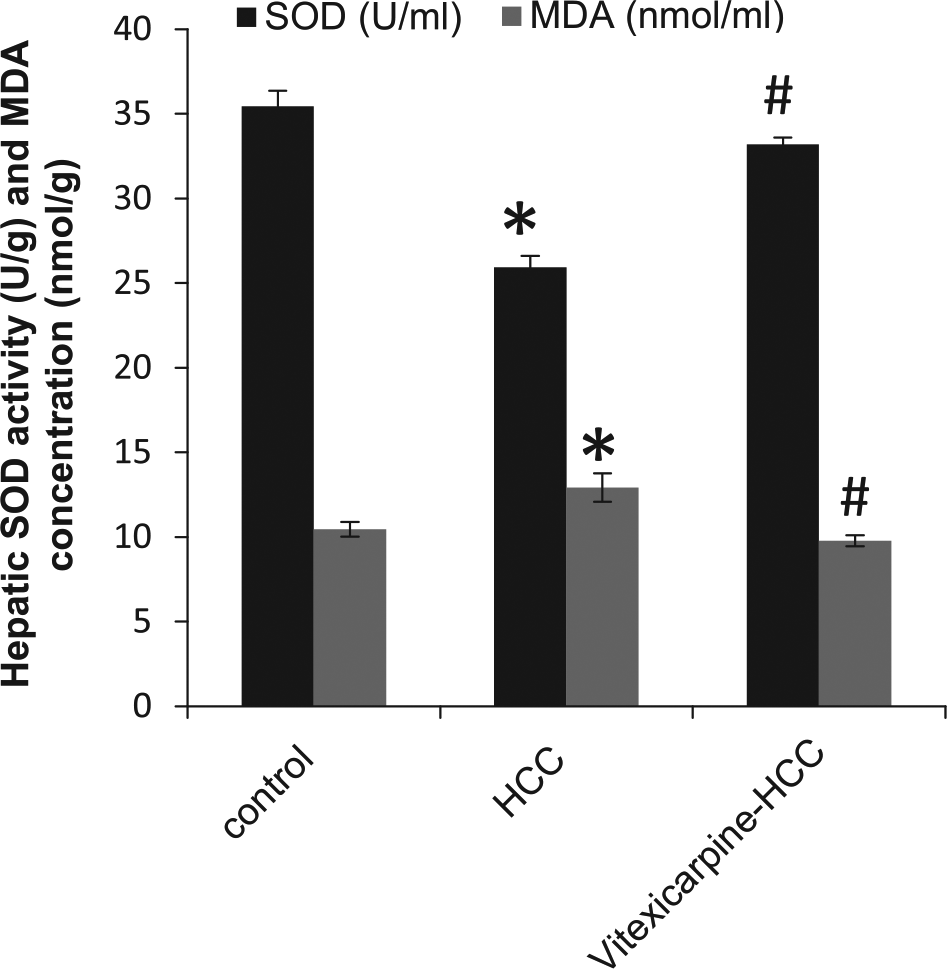
Hepatic measurements of superoxide dismutase (SOD, U/g) and malondialdehyde (MDA, nmol/g). A significant decrease in serum SOD activity and a significant increase in MDA in HCC group as compared with the control group. These effects were ameliorated by vitexicarpine.
Vitexicarpine blocked HCC-induced gene expression of total AKT
As illustrated in Figure 6, HCC group showed a significant increase in hepatic gene expression of total AKT (22.4-fold) compared with the control group. Treatment with vitexicarpine showed a strongly significant reduction in total AKT gene expression (0.08-fold) compared with HCC group, which reached a normal level.
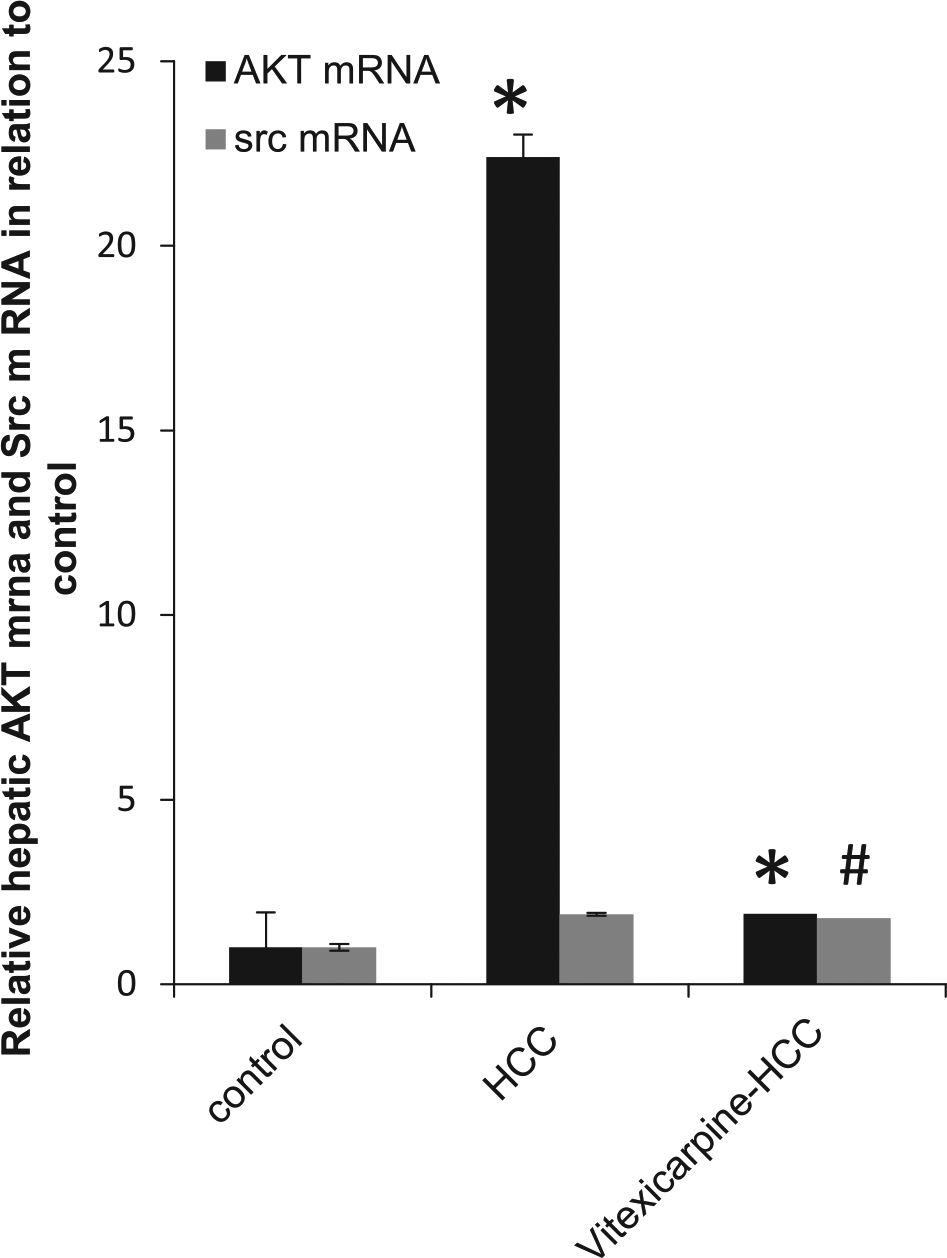
Relative gene expression of hepatic AKT mRNA and Src mRNA. HCC rats showed a significant increase in hepatic gene expression of total AKT and total Src as compared with the control group. Vitexicarpine normalized HCC-induced gene expression of total AKT.
Vitexicarpine reduced HCC-induced protein expression of p-AKT and p-Src
AKT and Src are activated by phosphorylation at Ser 473 and Tyr 419 sites, respectively. Accordingly, we studied the protein expression of phosphorylated AKT and Src by western blot using phospho-specific antibody against Ser 473 and Tyr 419, respectively. The expression p-AKT and p-Src was highly increased in HCC (5.9- and 8.8-fold, respectively) as compared with control group. While the treatment with vitexicarpine decreased the expression of p-AKT and p-Src (0.21- and 0.45-fold, respectively) as compared with HCC group (Figure 7). In addition, HCC showed a high increase in caspase-3 level (10-fold) as compared with the control group. However, treatment with vitexicarpine decreased caspase-3 level (0.41-fold) as compared with HCC group

Western blot analysis showed protein expression of caspase-3, phospho-Tyr419-Src (p-Src), phospho-Ser473-AKT (p-AKT), and β-actin in different studied groups. Vitexicarpine partially decreased in p-AKT, p-Src, and caspase-3 as compared with HCC group.
Immunohistochemistry for p-AKT and p-Src
Rat liver sections stained with p-AKT and p-Src antibodies showed increased p-AKT and p-Src protein content in hepatocytes that was reduced by treatment with vitexicarpine (Figure 8).

Immunohistochemical analysis of phospho-Ser 743-AKT (p-AKT) and phospho-Tyr419-Src (p-Src); both are partially decreased in vitexicarpine-treated rats as compared with HCC group.
Discussion
HCC is likely to produce angiogenic factors. VEGF is considered the strongest stimulator for angiogenesis, and it plays an important role in the course of carcinogenesis and metastasis.18,19 It is a well-established fact that VEGF is significantly elevated in HCC,20,21 as HCC is a highly vascular tumor, and tumor angiogenesis is critical for both its growth and metastasis.5,22,23
AKT is thought to be involved in survival pathways by inhibiting apoptotic processes. It plays an important role in multiple cellular processes such as cell proliferation, apoptosis, oxidative stress, and cell migration. 24 We found significant elevation in the expression of p-AKT/AKT in HCC, which agreed with previous studies.24–26 Many studies found a strong correlation between AKT activation and increased proliferation potential of HCC. 25
Src is closely associated with cancer progression and metastasis in humans. The HCC-induced activation of Src kinase correlates positively with cell proliferation, angiogenesis, motility, invasion, and migration.27,28 We found a significant increase in Src activation in HCC. Lu et al. 29 found that the overexpression of Src could increase p-ERK, p-AKT, VEGF, and JNK1 protein levels. Therefore, p-Src/Src activates hepatocarcinogenesis. Furthermore, the up-regulation of Src and its downstream pathways induce oncogenic transformation. The angiogenic response of VEGF was suggested to proceed via G-protein-coupled protein kinase AKT, Ras-independent focal adhesion kinase–Src–PI3K–Raf–MEK protein networks, and tyrosine kinase–regulated heregulin-activated signal pathways.30–35
Treatment with VEGF inhibitors markedly reduced the incidence of HCC in various studies.36–38 The identification of inhibitors of VEGF and VEGF-related protein kinases is an attractive approach toward developing new therapies for metastatic tumors and chronic inflammatory diseases. Vitexicarpine, a herbal medicine, was reported to inhibit VEGF-induced angiogenesis in vitro.39,40 Therefore, we examined vitexicarpine as a potentially novel anti-cancer agent in the treatment of HCC and tried to illustrate the pathways that may contribute to its angiogenic action. We found that treatment of HCC rats with vitexicarpine significantly reduced VEGF expression. This reduction in VEGF may have resulted by inhibiting the endothelial tube formation of cancer cells. 26 Furthermore, the significant decrease in gene expression of total AKT and phosphorylated AKT and Src by vitexicarpine treatment might be related to inhibition of VEGF. As AKT and Src play important role in angiogenesis, phosphorylation of them regulated VEGF signaling.26,29,41
Oxidative stress is highly involved in the development and progression of HCC. We found significant elevated MDA levels associated with reduced activity of SOD in HCC, which was reversed by vitexicarpine. These results are similar to the results of Kadir et al. 42 Antioxidant activity of vitexicarpine might be relevant to its free radical scavenging activity as well as the ability to release inducible nitric oxide synthase. 43 The reduction in the MDA levels is explained by the ability of vitexicarpine to avoid reactive oxygen species (ROS)-induced damage to membranes.44,45 The antiangiogenic activity of vitexicarpine also may be explained by consuming intracellular SODs, as well as, the mitochondrial-associated manganese-dependent SOD and the cytoplasmic/nuclear copper/zinc-dependent SOD toward triggering a burst of NOS drove the expression of cell suicide regulator and produced selective killing of tumor cell leading to an antiangiogenic effect. 10
Apoptosis plays a prominent part in the pathogenesis of HCC-induced liver injury. 46 Many previous studies suggest the role of caspase-3 in hepatocytes apoptosis.47,48 The presence of specific ligands can propagate the apoptosis signal by direct cleavage of downstream effect or caspases such as caspase-3. 49 Indeed, we found a significant increase in caspase-3 expression in HCC group, which was blocked by vitexicarpine. Zhu et al. 50 suggest that vitexicarpine is a potent apoptosis-inducing agent in human NSCLC cells, which acts through both pathways involving mitochondria and death receptor with subsequent activation of caspases.
The anticarcinogenic effect of vitexicarpine was revealed by the improvement of the survival rate of HCC rats and by the reduction in serum AFP. In addition, vitexicarpine produced hepatoprotective effect against HCC-induced liver damage, which resulted in the improvement of cell structure and the reduction in the activities of both ALT and GGT in HCC rats’ treatment with vitexicarpine.
Conclusion
Vitexicarpine produced antiangiogenic and anti-apoptotic activities in vivo by blocking VEGF signaling through the inhibition of both p-AKT/AKT and p-Src/Src. Therefore, vitexicarpine might be a good candidate for additional evaluation as a novel therapeutic agent for HCC.
Footnotes
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Ethical approval
The animal protocol was approved by the ethical committee of the Faculty of Pharmacy, Mansoura University, Mansoura, Egypt. All experiment procedures were conducted in compliance with the National Institutes of Health’s Guide for the Care and Use of Laboratory Animals (NIH publication no. 8023, revised 1978).
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the Faculty of Pharmacy, Mansoura University, Egypt.