
Abstract
Recently, we have demonstrated that IKBKE (inhibitor of nuclear factor kappa-B kinase subunit epsilon) is overexpressed in human glioblastoma and that inhibition of IKBKE remarkably suppresses the proliferative and invasive behaviour of glioblastoma cells. However, the specific pathogenic molecular mechanism remains to be elucidated. In this study, we verified that IKBKE promotes YAP1 expression via posttranslational modification and accelerates YAP1 translocation to the nucleus for the development of glioblastoma. We then determined that YAP1 negatively regulates miR-let-7b/i by overexpressing and silencing YAP1 expression. In addition, miR-let-7b/i feedback decreases the expression of IKBKE and YAP1 and suppresses the transportation of YAP1 located in the nucleus. Therefore, the regulatory feedback circuit of IKBKE↑→YAP1↑→miR-let-7b/i↓→IKBKE↑ dictates glioblastoma progression. Thus, we propose that blocking the circuit may be a new therapeutic strategy for the treatment of glioblastoma.
Introduction
Glioblastoma multiforme (GBM) is the most common and aggressive primary high-grade malignant glioma with high recurrence and morbidity rates. Although modern treatments with intracranial debulking surgery, radiotherapy plus concomitant and adjuvant temozolomide have been used for most patients, the prognosis of a median survival time of only 14.6 months is still fairly dreadful.1,2 Thus, elucidating the detailed molecular mechanisms and finding new therapeutic strategies have become the top priorities for curing glioblastoma.
IKBKE (inhibitor of nuclear factor kappa-B kinase subunit epsilon) is a non-canonical member of the IκB kinase (IKK) family. 3 Recent studies have shown that IKBKE was overexpressed in breast cancer, 4 glioma,5,6 ovarian cancer, 7 non–small cell lung cancer8,9 and renal clear cell carcinoma. 10 IKBKE expression was also closely related to the pathological grade of the cancer,6,7,9 chemoresistance7,8 and induced malignant transformation.4,9 Li et al. 6 have demonstrated that the knockdown of IKBKE inhibits proliferation, migration and invasion in vitro and suppresses glioblastoma tumourigenesis in vivo. These data show that IKBKE has an important role in glioblastoma progression, but its specific mechanism remains unknown.
YAP1, a momentous downstream factor of the Hippo pathway, acts as a vital transcription co-activator to regulate cell stemness, proliferation, differentiation, apoptosis and death. 11 Current research has shown that elevated YAP1 expression as an oncogene12,13 was found in multiple types of human cancer, such as oesophageal squamous cell carcinoma, 14 cervical carcinoma, 15 hepatocellular carcinoma, 16 non–small cell lung cancer 17 and glioblastoma. 18 Orr et al. 18 showed that YAP1 was overexpressed in high-grade gliomas, especially in anaplastic astrocytoma (The World Health Organization (WHO) grade III) and glioblastoma (WHO grade IV). The silencing of YAP1 may significantly inhibit the proliferation of glioblastoma cell lines, 18 demonstrating that YAP1 has a significant effect on glioblastoma progression.
The miR-let-7 family was identified as a vital tumour suppressor in numerous types of cancer. 19 Recent studies demonstrated that let-7 could inhibit glioblastoma cell growth and migration through various pathways.20–22 Tian et al. 21 reported that let-7b/i, which was downregulated in glioblastoma cell lines, could inhibit glioblastoma cell migration and invasion by directly targeting IKBKE, affecting epithelial–mesenchymal transition (EMT) in glioma cell lines. Therefore, let-7b/i may become a new therapeutic target for glioblastoma.
For this article, we used two representative glioblastoma cell lines, U87-MG and LN-229, as investigative subjects. We show that silencing IKBKE suppresses the expression of total YAP1, thus increasing the expression of miR-let-7b/i. The elevated miR-let-7b/i further decreased IKBKE expression, thereby establishing the regulatory feedback loop of IKBKE↑→YAP1↑→miR-let-7b/i↓→IKBKE↑ to promote glioblastoma progression.
Materials and methods
Cell culture
Human glioblastoma cell lines, U87-MG and LN-229, were purchased from the Institute of Biochemistry and Cell Biology, Chinese Academy of Sciences, Shanghai, China. The cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM, Gibco, USA) supplemented with 10% foetal bovine serum (FBS; Gibco) and incubated at 37°C with 5% CO2.
Cell transfection with IKBKE–short hairpin RNA, YAP1–small interfering RNA, miR-let-7b/i mimics and IKBKE, Flag-YAP1 plasmids
We established an IKBKE–short hairpin RNA (shRNA) lentiviral vector (Shanghai GeneChem, Shanghai, China) using the sequence: 5′-GCATCATCGAACGGCTAAATA-3′. A green fluorescent protein (GFP) scrambled lentiviral vector with the sequence: 5′-TTCTCCGAACGTGTCACGTTTC-3′ was used as the negative control (NC). The designs of YAP1–small interfering RNA (siRNAs) were from Ribobio (Guangzhou, China). The oligonucleotide sequence of the miR-let-7b mimics was 5′-UGAGGUAGUAGG UUGUGUGGUU-3′, and sequence of the miR-let-7i mimics was 5′-UGAGGU AGUAGUUUGUGCUGUU-3′. A scrambled microRNA sequence (5′-UUCUCC GAACGUGUCACGUTT-3′) was used as the NC. And the transfection process of microRNAs and siRNAs were performed as previously described. 6 The IKBKE and Flag-YAP1 plasmid and their vector plasmids were purchased from Addgene (USA). We selected Lipofectamine 3000 from Thermo Fisher Scientific (USA) as transfection reagent and the process of transfection was followed by the manufacturer’s instructions.
RNA extraction and real-time polymerase chain reaction analysis
The total RNA of the glioblastoma cell lines was extracted with TRIzol (Invitrogen, USA) and reverse transcription was performed using the GoScriptTM Reverse Transcription System (Promega, USA) for complementary DNA (cDNA) synthesis following the manufacturer’s protocol. Then, quantitative real-time polymerase chain reaction (PCR) was performed using the quantitative PCR (qPCR) Mix System (Promega) according to the manufacturer’s instructions. The reaction conditions were as follows: 95°C for 5 min and 40 cycles of 95°C for 12 s and 60°C for 40 s. The nucleotide sequences of the primers were as follows: glyceraldehyde 3-phosphate dehydrogenase (GAPDH): 5′-GGAGCGAGATCCCTCCAAAAT-3′ (forward primer) and 5′-GGCTGTTGTCATACTTCTCATGG-3′ (reverse primer); IKBKE: 5′-GAGAAGTTCGTCTCGGTCTATGG-3′ (forward primer) and 5′-TGCATGGTACAAGGTCACTCC-3′ (reverse primer); YAP1: 5′-TAGCCCTGCGTAGCCAGTTA-3′ (forward primer) and 5′-TCATGCTTAGTCCACTGTCTGT-3′ (reverse primer); GAPDH was used as the control. The reverse transcriptional primers and PCR primers for miR-let-7b/i were synthesized by Ribobio. U6 was used as the internal control for miR-let-7b/i real-time PCR and the 2−ΔΔCT method was used to analyse PCR results.
Protein extraction and western blot analysis
Total cell protein was extracted as previously described. 6 The cytoplasmic and nuclear proteins were extracted using the nuclear and cytoplasmic protein extraction kit from Beyotime (Guangzhou, China). The protein concentration was measured using the bicinchoninic acid (BCA) protein assay kit (Solarbio, Beijing, China). Then, 20 mg of protein was mixed with 4× sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) loading buffer, loaded into each lane, separated by gel electrophoresis using 10% or 12% SDS-PAGE gels and transferred to polyvinylidene fluoride (PVDF) membranes (Millipore, USA). After being washed three times with Tris-buffered saline with Tween 20 (TBST), the membrane was blocked with 5% bovine serum albumin (BSA) for 1 h at 37°C. Then, the membrane was incubated with primary antibodies overnight at 4°C. After being rewarmed for 1 h, the membrane was washed three times before incubation with a secondary antibody (1:2000, Beijing, China) for 1 h at room temperature and was washed three times. The protein bands were detected using the G:BOX (Syngene Company, UK) with Chemiluminescent horseradish peroxidase (HRP) Substrate (Millipore).
Immunofluorescence
Different cell lines were seeded onto sterile cover slips in a 12-well plate overnight. Cells were washed with phosphate-buffered saline (PBS) and fixed with 4% paraformaldehyde for 20 min. Then, cells were permeabilized with 0.1% Triton X-100 for 15 min and blocked with 5% BSA at 37°C for 30 min. Cells were then incubated with primary antibody at 4°C overnight. After being rewarmed for 1 h, samples were washed and incubated with specific secondary antibodies (1:200; Thermo Fisher Scientific) for 2 h at 37°C. After washing three times, nuclei were stained with DAPI (4′,6-diamidino-2-phenylindole) for 5 min at room temperature. Immunofluorescence was measured using confocal microscopy after sealing the cells with an anti-quenching reagent.
Cell Counting Kit-8 assay
Cells were seeded (3000/100 µL/well) into 96-well plates. From the first day to the fifth day, 10 µL of Cell Counting Kit-8 (CCK-8) reagent (Dojindo, Japan) was added into each well and incubated for 3 h at 37°C every day. Then, the 96-well plate OD value was measured using a microplate reader at 450 nm.
Cell migration and invasion assay
The transwell assay without matrix was used to assess cells migration. Cells were seeded (5 × 104/200 µL) into transwell chambers with serum-free DMEM. The outer chambers were filled with 500 µL 10% FBS DMEM. Cell invasion was estimated using the transwell assay with Matrigel. Before cells were seeded into chambers, Matrigel with three times the volume (60 µL) of serum-free DMEM was coated on the upper surface of the chamber. Then, the chambers were incubated at 37°C for 30 min, to allow for Matrigel solidification. After incubation for 24 h (migration) or 48 h (invasion), cells attached to the bottom side of the membrane were fixed with 4% paraformaldehyde for 15 min, stained with crystal violet for 1 min, counted and imaged under the microscope.
Statistical analysis
We used the SPSS software (version 16.0) for statistical analysis. All data are shown as the mean ± standard deviation (SD). A value of p < 0.05 was considered to be statistically significant.
Results
Inhibition of IKBKE suppresses proliferation, migration and invasion in glioblastoma cells
First, we silenced IKBKE expression using shRNA-IKBKE in U87-MG and LN-229 cells, and the inhibitory effect of IKBKE was determined by western blot and real-time PCR (Figure 1(a) and (b)). Then, the CCK-8 assay was used to assess proliferation in cells transfected with IKBKE-shRNA and scrambled shRNA. As shown in Figure 1(c), U87-MG and LN-229 cells infected with IKBKE-shRNA lentivirus grew noticeably slower than cells transfected with scrambled shRNA (NC) and blank control groups. To determine whether IKBKE had an impact on the migration and invasion of glioblastoma cells, we used the transwell assay with or without Matrigel (Figure 1(d) and (e)). Across the transwell chamber, the average number of both migratory and invasive cells with IKBKE knockdown was much less than the blank control and NC groups (Figure 1(d) and (e)). These data demonstrate that the suppression of IKBKE could inhibit the proliferative, migratory and invasive abilities of glioblastoma cells.

IKBKE knockdown suppresses proliferation, migration and invasion in U87-MG and LN-229 cells. (a) IKBKE knockdown was determined by western blot. (b) IKBKE knockdown was determined by real-time PCR. (c) The CCK-8 assay was used to assess cell proliferation after inhibition of IKBKE. (d) Transwell assay without Matrigel for assessing the migration of cells after IKBKE knocked down. (e) Transwell assay coated with Matrigel for assessing the invasion of cells after IKBKE knocked down.
IKBKE increases YAP1 expression, transports YAP1 to the nucleus and inhibits miR-let-7b/i expression
To further investigate the effect of IKBKE on YAP1 expression, we demonstrated that YAP1 expression decreased and p-YAP1 (s127) expression increased compared to the blank control and NC groups by western blot (Figure 2(a)). As is known, YAP1 is directly phosphorylated by LATS1/2 at Ser127, causing its sequestration in the cytoplasm and then degradation through ubiquitylation by 14-3-3 under normal conditions.11,23 So our data indicated that inhibition of IKBKE could increase YAP1 degradation. However, interestingly, the messenger RNA (mRNA) of YAP1 barely changed as measured by real-time PCR after IKBKE knockdown (Figure 2(b)). Meanwhile, miR-let-7b and miR-let-7i expression increased compared to the blank control and NC groups (Figure 2(b)), indicating that the downregulation of IKBKE can decrease YAP1 expression and increase miR-let-7b/i expression.

IKBKE increases YAP1 expression, transports YAP1 to the nucleus and inhibits miR-let-7b/i expression. (a) Inhibition of IKBKE increased p-YAP1 (s127) and decreased YAP1 expression by western blot. (b) Inhibition of IKBKE increased miR-let-7b/i expression, but barely changed the expression of YAP1 mRNA. (c) IKBKE knockdown inhibited YAP1 transposition into the nucleus using western blot. (d) IKBKE knockdown inhibited YAP1 transposition into the nucleus by fluorescence analysis. (e) Overexpressed IKBKE increased YAP1 expression and decreased p-YAP1 (s127) by western blot. (f) IKBKE and YAP1 mRNA levels were measured by real-time PCR after overexpressing IKBKE. (g) Overexpressed IKBKE decreased miR-let-7b/i expression.
YAP1 with an unphosphorylated Ser127 resulted from an ineffective Hippo pathway and was translocated to the nucleus, forming transcription co-activators to induce transcription of specific genes. 24 To determine whether IKBKE affects YAP1 translocation activity, we studied YAP1 expression in the cytoplasm and nucleus after IKBKE knockdown in U87-MG and LN-229 cells. As shown in Figure 2(c), YAP1 expression in the nucleus was markedly decreased after silencing IKBKE compared to the blank control and NC groups, while cytoplasmic YAP1 increased after treatment with shRNA-IKBKE. These data were further confirmed by fluorescence analysis (Figure 2(d)), suggesting that silencing of IKBKE inhibits total YAP1 expression, prevents YAP1 nuclear translocation and increases miR-let-7b/i expression.
To further investigate the IKBKE effect on YAP1 and miR-let-7b/i, we then overexpressed IKBKE using an IKBKE plasmid. First, we determined the efficiency of overexpressed IKBKE using western blot (Figure 2(e)) and real-time PCR (Figure 2(f)). The expression of total YAP1 increased and the expression of p-YAP1 (s127) decreased after overexpressing IKBKE compared to the blank control and NC groups (Figure 2(e)), while the change in the mRNA of YAP1 was negligible (Figure 2(f)). Meanwhile, miR-let-7b and miR-let-7i expression decreased after transfection with IKBKE plasmid compared to the blank control and NC groups using real-time PCR (Figure 2(g)). Together, the data demonstrate that IKBKE overexpression increased the total YAP1 expression and decreased miR-let-7b/i expression.
YAP1 reduces miR-let-7b and miR-let-7i expression
To investigate whether silencing of IKBKE increased miR-let-7b/i expression by decreasing YAP1, we overexpressed YAP1 after IKBKE knockdown using a YAP1 plasmid. The effect of YAP1 overexpression was determined by western blot (Figure 3(a)) and real-time PCR (Figure 3(b)). After transfection with YAP1 plasmid in glioblastoma cells that were previously infected with shRNA-IKBKE lentivirus, we measured miR-let-7b and miR-let-7i expression separately using real-time PCR. As shown in Figure 3(c), miR-let-7b and miR-let-7i expression first increased after transfection with shRNA-IKBKE and then decreased as YAP1 was overexpressed, demonstrating that YAP1 overexpression can reverse the increased expression of miR-let-7b and miR-let-7i caused by IKBKE knockdown.

YAP1 reduces miR-let-7b/i expression. (a) Overexpression of YAP1 was determined by western blot. (b) YAP1 mRNA level was upregulated by real-time PCR after overexpressing YAP1. (c) Overexpression of YAP1 decreased miR-let-7b/i expression. (d) Knockdown of YAP1 using siRNAs was determined by western blot. (e) Knockdown of YAP1 using siRNAs was determined by real-time PCR. (f) Knockdown of YAP1 increased miR-let-7b/i expression.
Next, we silenced YAP1 expression using siRNAs in glioblastoma cells. We tested the ability of three siRNAs to silence YAP1. Real-time PCR analysis of YAP1 expression after transfection with siRNA for 48 h (Figure 3(d)) showed that siR-3 was the most efficient siRNA to knock down YAP1 compared to NC group. Furthermore, knockdown of YAP1 was confirmed at the protein level by western blot (Figure 3(e)). Thus, we used siR-3 to silence YAP1 in the following experiments. We found that miR-let-7b and miR-let-7i were increased compared to the blank control and NC groups after silencing YAP1 using siR-3 (Figure 3(f)), demonstrating that YAP1 downregulation increases miR-let-7b/i.
MiR-let-7b and miR-let-7i inhibit IKBKE and YAP1 expression
Previous work demonstrated that miR-let-7b/i suppressed glioblastoma cell line progression by directly targeting IKBKE to decrease IKBKE expression. 21 Therefore, we overexpressed miR-let-7b and miR-let-7i using respective mimics to investigate its function on IKBKE and YAP1. As shown in Figure 4(a), the overexpression efficiency of miR-let-7b and miR-let-7i in cells was measured by miRNA quantitative real-time PCR and the mimics were overexpressed compared to the blank control and NC groups. Then, the expression of IKBKE and total YAP1 was determined by western blot (Figure 4(b)), showing that IKBKE and total YAP1 expression were significantly decreased after separate transfection with miR-let-7b and miR-let-7i mimics.
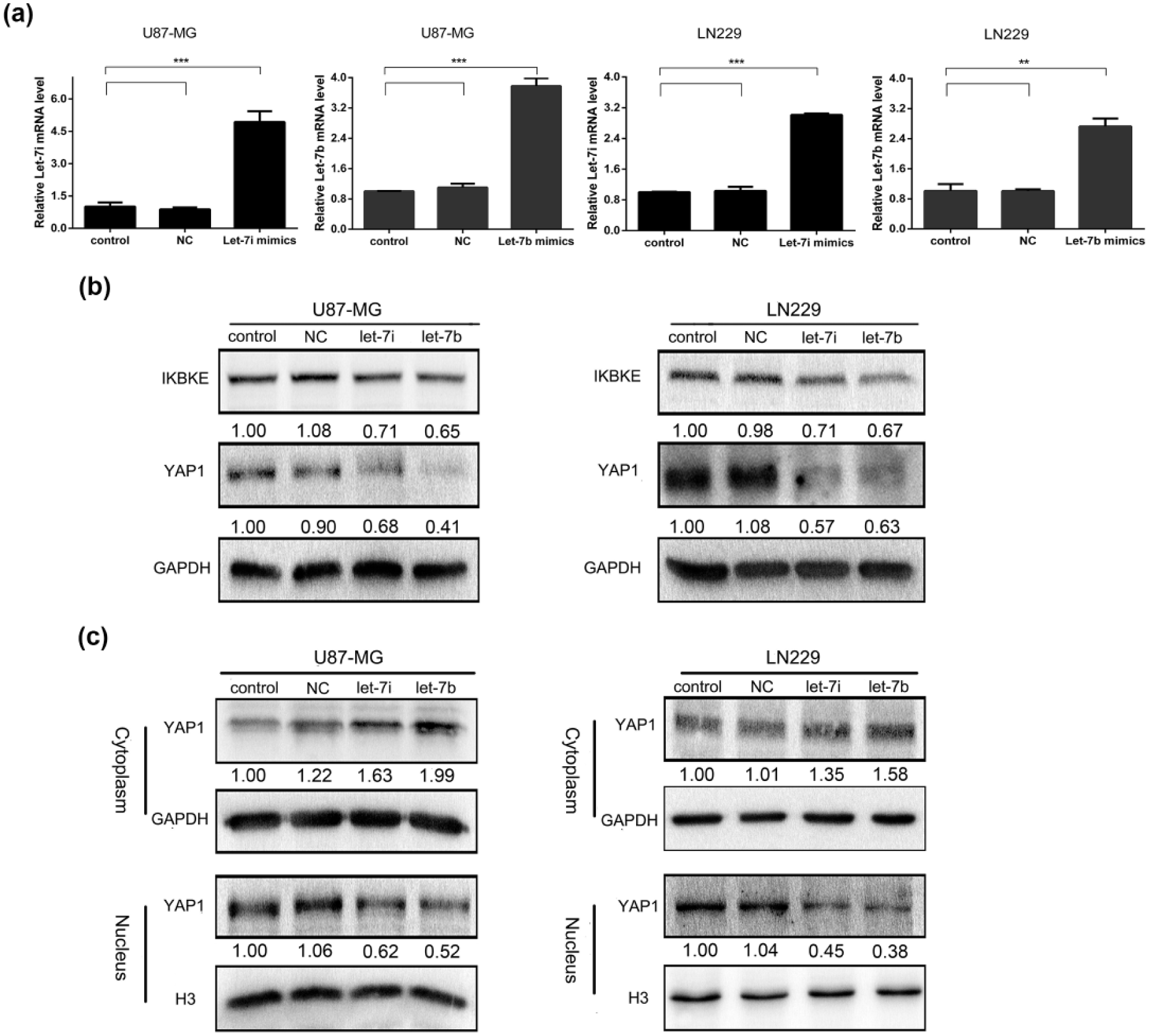
miR-let-7b/i inhibit IKBKE and YAP1 expression. (a) Overexpression of miR-let-7b/i using mimics was determined by teal-time PCR. (b) MiR-let-7b/i downregulated IKBKE and YAP1 expression by western blot. (c) MiR-let-7b/i inhibited YAP1 transposition into the nucleus using western blot.
To further explore that the effect of miR-let-7b/i overexpression on YAP1 translocation, we analysed YAP1 expression in the cytoplasmic and nuclear extracts of transfected glioblastoma cells using western blot. As shown in Figure 4(c), after transfection with miR-let-7b and miR-let-7i mimics, YAP1 expression in the nucleus clearly decreased compared to the blank control and NC groups, while the corresponding cytoplasmic YAP1 expression noticeably increased. These data suggest that miR-let-7b/i inhibits the expression of IKBKE and total YAP1 and attenuates YAP1 translocation to the nucleus to suppress glioblastoma progression.
Discussion
Recent studies have demonstrated that IKBKE is upregulated in glioblastoma, where it controls proliferation, migration and invasion in vitro and promotes tumorigenesis in vivo.5,6 Pathways involving IKBKE are well understood to promote oncogenesis, embrace the nuclear factor-κB (NF-κB) pathway,25–27 phosphoinositide 3-kinase/Akt/mammalian target of rapamycin (PI3K/Akt/mTOR) pathway28,29 and so on. In this study, we discovered that IKBKE increased the expression of YAP1, an important downstream factor of the Hippo pathway and advanced YAP1 translocation to the nucleus to accelerate glioblastoma progression. Remarkably, with IKBKE knockdown or overexpression, the change in YAP1 mRNA expression was negligible, while YAP1 protein expression changed with IKBKE expression. Thus, suggesting that IKBKE regulates YAP1 expression mainly through posttranslational modification rather than at a transcriptional level. By measuring the change in YAP1 expression in the cytoplasm and nucleus by western blot, we determined that IKBKE inhibition could decrease YAP1 translocation to the nucleus to depress YAP1-dependent transcription. Therefore, IKBKE may attenuate Hippo pathway phosphorylation of YAP1 through other regulatory mechanisms to weaken YAP1 degradation in the cytoplasm and promote YAP1 transcriptional activity in the nucleus, thus strengthening malignancy.
Previous studies have demonstrated that YAP1 acted as a crucial transcription factor whose transcriptional activity was mainly suppressed by the Hippo pathway through a combination of SMADs, TEADs forming co-activators to induce target genes transcription.30,31 Recent studies simultaneously showed that miR-let-7 was mainly regulated by LIN28, an important RNA binding protein, targeting pre-let-7 to block its maturity, thus facilitating carcinogenesis.32–34 Meanwhile, Chaulk et al. 35 recently showed that YAP1 successfully decreased mature let-7 expression by regulating the LIN28/Let-7 axis in breast cancer cell lines. Interestingly, YAP1 inhibition did not decrease mRNA expression of LIN28 but markedly decreased LIN28 protein expression, indicating that YAP1 may regulate LIN28 through post-transcriptional events, therefore, to reduce mature let-7 expression with no effect on pre-let-7. Our data further demonstrated this conclusion in glioblastoma cell lines by measuring the change in mature let-7b/i expression after silencing or overexpressing YAP1. Given that IKBKE increased the expression of YAP1 and led to its translocation to the nucleus, it was reasonable that IKBKE indirectly decreased miR-let-7b/i expression by regulating YAP1 expression.
Early studies demonstrated that the miR-let-7 family acted as tumour suppressors by suppressing several pivotal regulators in cancer pathways. Johnson et al. 36 demonstrated that ras genes, including H-ras, K-ras and N-ras, were negatively regulated by miR-let-7 to modulate a series of cell processes. Shi et al. 37 reported that HMGA (high-mobility group A), a critical oncogene, was a downstream target of miR-let-7 in uterine leiomyosarcoma. He et al. 38 determined that miR-let-7a suppressed the growth of lung cancer cells by targeting NIRF. Schultz et al. 39 showed that miR-let-7b affects the cell cycle by directly targeting CCND1 in malignant melanoma. Wang et al. 40 also reported that miR-let-7a inhibited STAT3 expression to facilitate hepatocarcinogenesis. Our laboratory recently reported that miR-let-7b/i markedly suppressed migration and invasion in glioblastoma cell lines by directly targeting IKBKE. 21 Together, these studies show that the miR-let-7 family plays an important role in malignancy by inhibiting crucial oncoproteins. Our data confirm that the miR-let-7b/i inhibitory effect on IKBKE suppresses YAP1 expression and YAP1 translocation to the nucleus.
In conclusion, we determined that IKBKE knockdown decreased total YAP1 expression on a protein level rather than on a mRNA level, inhibiting YAP1 translocation to the nucleus and increasing miR-let-7b/i expression, while IKBKE overexpression inversed the outcomes. Then, we validated that YAP1 suppressed miR-let-7b/i by measuring miR-let-7b/i after silencing and overexpressing YAP1. Furthermore, overexpressing miR-let-7b/i reduced IKBKE and total YAP1 expression, decreased YAP1 accumulation in the nucleus and consequently inhibited YAP1 transcription similar to IKBKE knockdown. Therefore, we constructed the regulatory feedback loop of IKBKE↑→YAP1↑→miR-let-7b/i↓→IKBKE↑ and proposed that the loop would be a vital way to promote glioblastoma development and that obstructing the circuit might be a novel and helpful therapeutic strategy for glioblastoma treatment.
Footnotes
Acknowledgements
Z.Z., J.L. and G.G. contributed equally to this work.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work is supported by the National Nature Science Foundation of China, Grant No. 81572490.