
Abstract
Multiple myeloma is the most common cause of death of hematological malignancy worldwide. Cullin 4A has been proposed as oncogene in several types of human cancer, but the expression and function of cullin 4A in multiple myeloma remain unclear. Here, we demonstrate that cullin 4A plays an oncogenic role in multiple myeloma development. The expression of cullin 4A was detected by quantitative real-time polymerase chain reaction in multiple myeloma patients and multiple myeloma cell lines. In addition, silencing of cullin 4A with small interfering RNA was performed in human multiple myeloma cells, and the impact on proliferation, cell cycle, apoptosis, migration, and invasion of the multiple myeloma cells was analyzed. We found that the level of cullin 4A in serum samples was significantly upregulated in patients with multiple myeloma compared with healthy control subjects. Knockdown of cullin 4A via small interfering RNA inhibited the proliferation of the multiple myeloma cell lines by delaying cell-cycle progression and increasing apoptosis. cullin 4A downregulation inhibited multiple myeloma cell migration and invasion in vitro. Our results suggested that cullin 4A could be a promising therapy target in multiple myeloma patients.
Multiple myeloma (MM) is the second most common hematological cancer worldwide and is characterized by the clonal proliferation of neoplastic serum cells in the bone marrow (BM). 1 The major symptoms of MM are hypercalcemia, anemia, renal insufficiency, bone lesions (CRAB), and susceptibility to infection. 2 Although current therapies may improve the patient’s survival, MM remains an incurable disease for the vast majority of patients. 3 Thus, novel treatment strategies to effectively control MM still needs to be resolved.
Cullin 4A (CUL4A) is an 87-kDa protein, which belongs to the family of evolutionally conserved cullin proteins that composes the multifunctional ubiquitin ligase E3 complex, and is essential for the ubiquitination of several well-defined tumor suppressor genes.4,5 CUL4A is involved in the control of cell cycle, DNA replication, and DNA repair depending on the nature of ubiquitinated proteins.6,7 CUL4A has been found amplified and/or overexpressed in different types of carcinomas and a putative oncogenic role has been proposed for this gene.8,9 However, the precise function of CUL4A in human MM has never been reported. In this study, we attempted to elucidate the potential role of CUL4A in growth and tumorigenesis of MM. Depletion of CUL4A by short hairpin RNA (shRNA) inhibited proliferation, led to mesenchymal-to-epithelial transition (MET), and blocked distant metastasis. These collectively established data highlight CUL4A as a potential oncogenic biomarker and therapeutic target for MM patients.
Materials and methods
Serum samples
A total of 68 serum samples were obtained from patients with MM who underwent treatment at Jiulongpo District People’s Hospital between 2010 and 2015 (42 cases with κ type, 20 cases with λ type, and 6 cases with non-secretory type). There were 40 serum samples from the normal control without any detectable BM abnormalities. The diagnostic criteria for all MM patients were determined by International Myeloma Working Group 2012 system. This study was approved by the Ethics Committee of Jiulongpo District People’s Hospital. All patients provided informed consent.
Cell lines
The human MM cell lines U266, KM3, and RPMI 8226 were obtained from American Type Culture Collection (Manassas, VA, USA). All cells were maintained in RPMI 1640 medium (Gibco; Thermo Fisher Scientific, Inc., St. Louis, MO, USA) supplemented with 10% fetal bovine serum (FBS; Gibco; Thermo Fisher Scientific, Inc.) at 37°C in a humidified atmosphere containing 5% CO2. The cells at the logarithmic growth phase were harvested for the subsequent experiments when the cells reached 80% confluence.
Quantitative real-time polymerase chain reaction
Total RNA was extracted from cells using RNAiso Plus reagent (TaKaRa, Dalian, China), and complementary DNA (cDNA) was synthesized using PrimeScript RT Reagent (TaKaRa). Quantitative real-time polymerase chain reaction (qRT-PCR) was performed on a StepOnePlus Real-Time PCR System (Applied Biosystems, Carlsbad, CA, USA) according to the manufacturer’s instructions. The PCR conditions consisted of an initial denaturation step at 95°C for 10 min, followed by 40 cycles of 15 s at 95°C and 1 min at 60°C. Finally, a melting curve profile was set at 95°C (15 s), 60°C (15 s), and 95°C (15 s). Each messenger RNA (mRNA) level was measured as a fluorescent signal corrected according to the signal for glyceraldehyde 3-phosphate dehydrogenase (GAPDH). The sequences of the primer pairs are follows: CUL4A forward, 5′-ATACTTCAGGACCCACGTTTGAT-3′, CUL4A reverse, 5′-TCTCCAAGTACTAAAGCAGGAAAATCT-3′; GAPDH forward, 5′-ATAGCACAGCCTGGATAGCAACGTAC-3′, GAPDH reverse, 5′-CACCTTCTACAATGAGCTGCGTGTG-3′.
Western blot analysis
Cells were harvested and lysed in radioimmunoprecipitation assay (RIPA) buffer and protease inhibitor cocktail (Roche Applied Science, Foster City, CA, USA). Protein concentration of samples was determined by detergent-compatible colorimetric assay (Bio-Rad Laboratories, Hercules, CA, USA). A volume of 20µg of protein was subjected to electrophoresis on NuPAGE 4% to 12% Bis–Tris gels and MOPS running buffer (Novex, Carlsbad, CA, USA) followed by blotting to nitrocellulose membranes. The blots were analyzed using polyclonal rabbit anti-CUL4A (1:1000; Cell Signaling Technology, Danvers, MA, USA), polyclonal rabbit anti-E-cadherin, polyclonal rabbit anti-N-cadherin, polyclonal rabbit anti-vimentin (Santa Cruz Biotechnology, Santa Cruz, CA, USA), and monoclonal mouse anti-β-actin (1:12,000; Sigma-Aldrich, St. Louis, MO, USA) antibodies. The immunoblot signals were quantified using the ImageJ 1.43u software (Wayne Rasband, National Institutes of Health). CUL4A bands’ intensity was normalized to β-actin bands.
RNA interference
Cells were seeded in a six-well plate as 50,000 cells/well with fresh media without antibiotics 24 h before transfection, with a target of 30%–50% confluency at the time of transfection. CUL4A-siRNA (ON-TARGETplus SMARTpool) and control siRNA were purchased from Thermo Fisher Scientific (Waltham, MA, USA). Cells were transfected with 100 nmol/L of siRNA using Lipofectamine RNAiMAX (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s protocol. After siRNA transfection, the plates were incubated for 48 h at 37°C before further analysis.
Cell proliferation assay and cell-cycle analysis
Cells were seeded in 96-well plates at 1 × 103 per well. Cell proliferation was evaluated using Cell Counting Kit-8 (CCK-8; Dojindo, Rockville, USA) according to the manufacturer’s instructions. Briefly, 10 µL of CCK-8 solution was added to culture medium and incubated for 2 h. The absorbance at 450 nm wavelength was determined with a reference wavelength of 570 nm. For cell-cycle analysis, cells were plated in six-well plates at 5 × 105 per well. The cell-cycle distribution was analyzed by propidium iodide (Sigma-Aldrich) staining and flow cytometry. All experiments were performed in triplicates.
Colony-formation assay
Cells were plated into three 6-cm cell culture dishes after transfected with CUL4A-siRNA or control siRNA. Cells were incubated for 2 weeks in complete growth media. Cell colonies were fixed with cold methanol stained with 0.1% crystal violet for 30 min. The colonies were manually counted using a microscope.
Apoptosis analysis
At transfection, cells were harvested and washed with ice-cold phosphate-buffered saline twice. Then, cells were re-suspended in Annexin V Binding Buffer and the indicated amount of propidium iodide and Annexin V–fluorescein isothiocyanate (FITC; BD Pharmingen, San Diego, CA, USA) was added. Cells were analyzed by flow cytometry (FACSCalibur; BD Biosciences, Franklin Lakes, NJ, USA). The proportion of apoptotic cells (Annexin V positive cells) was shown as the mean ± standard deviation (SD).
Cell migration and invasion assays
Cell culture was performed in Transwell chambers (8-µm pore size; Corning Costar, Corning, NY, USA). For the migration assay, 5 × 104 cells were plated into 200 µL RPMI 1640 medium in the upper chamber and were cultured for 36 h. For the invasion assay, the insert membranes were coated with diluted Matrigel (BD Biosciences, San Jose, CA, USA), but were cultured under the same conditions. Finally, the membranes were removed and stained with hematoxylin, and images were captured using an inverted microscope (Olympus BX51; Olympus Corporation, Tokyo, Japan).
Statistical analysis
All statistical analyses were carried out with the SPSS statistical software package (version 16.0; SPSS, Inc., Chicago, IL, USA). The Student’s t-test was used for comparisons. All p values were two-sided and p values less than 0.05 was considered to be statistically significant.
Results
CUL4A is overexpressed in MM serum and cell lines
The expression of CUL4A in the serum of 68 patients with MM and 40 normal donors was examined by qRT-PCR. It was found that the level of CUL4A in serum samples was significantly upregulated in patients with MM compared with healthy control subjects (p < 0.001, Figure 1(a)), indicating that CUL4A may contribute to the development of MM. In addition, the expression level of CUL4A in MM cell lines was evaluated. CUL4A was significantly increased with different levels in all of these MM cell lines, compared with normal serum cells from healthy donors (Figure 1(b)). In addition, the highest level of CUL4A was found in U266 and RPMI 8226. U266 and RPMI 8226 cell lines were used as the research model in the rest of the experiments.
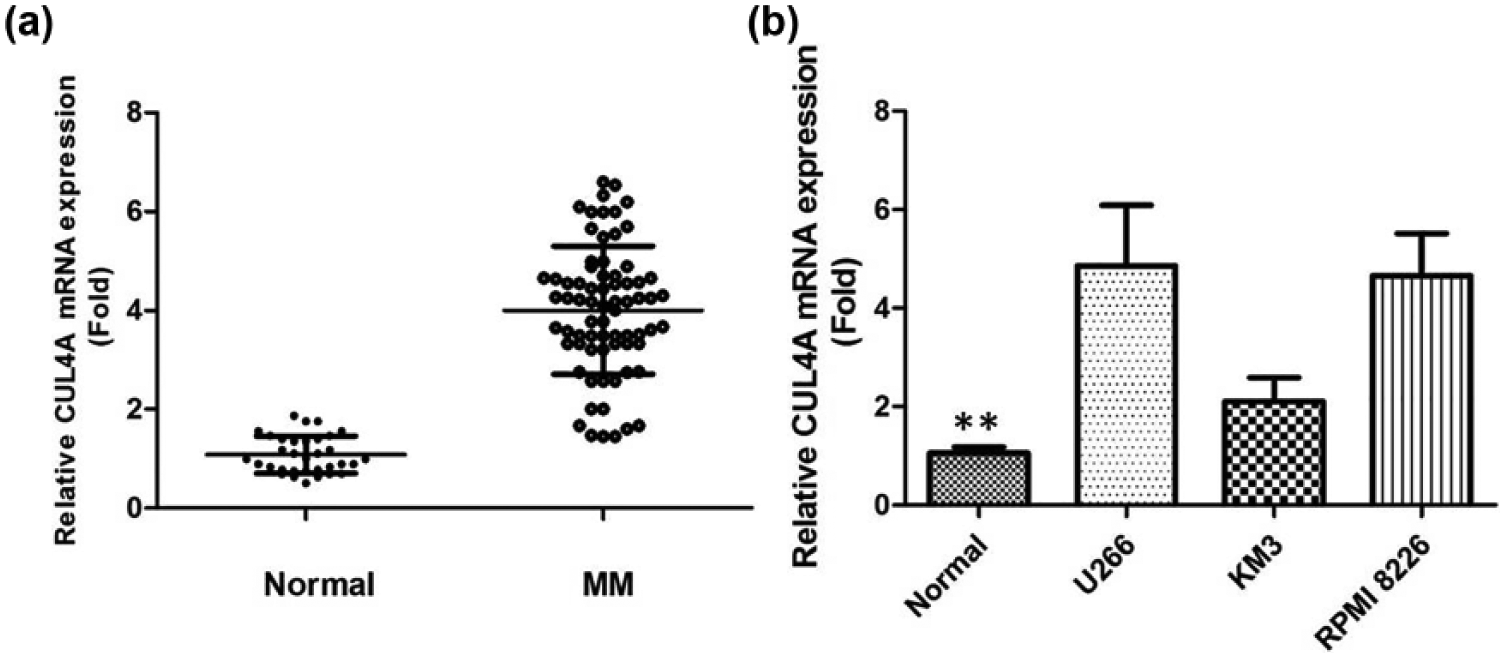
(a) Quantitative real-time PCR shows expression level of CUL4A mRNA in patients with MM. (b) Quantitative real-time PCR shows expression level of CUL4A mRNA in MM cell lines.
Inhibition of CUL4A inhibits cell proliferation in MM cell lines
To investigate the role of CUL4A in MM progression, we performed a loss-of-function experiment by knocking down CUL4A expression using specific siRNA. Using qRT-PCR and Western blotting, we found significant suppression of CUL4A gene expression in transfected in U266 and RPMI 8226 cells, suggesting the silencing of endogenous gene expression by siRNA (Figure 2; p < 0.01). We investigated the proliferation activity by CCK-8 assay in vitro. The results showed that the growth rate of all U266 and RPMI 8226 cells transfected by CUL4A-siRNA was slower than control cells (Figure 3(a) and (b); p < 0.01). Similarly, the results of colony-formation assays revealed that U266 and RPMI 8226 cells following inhibition of CUL4A had significantly lower number of colony formation compared with control, suggesting that CUL4A may act as an oncogene involved in the promotion of MM cell proliferation (Figure 3(c) and (d); p < 0.01). CUL4A knockdown induced G2- and S-phase cell-cycle arrest: the percentage of cells in G2 and S phases was higher in CUL4A-siRNA U266 and RPMI 8226 than control (Figure 4(a) and (b); p < 0.01).
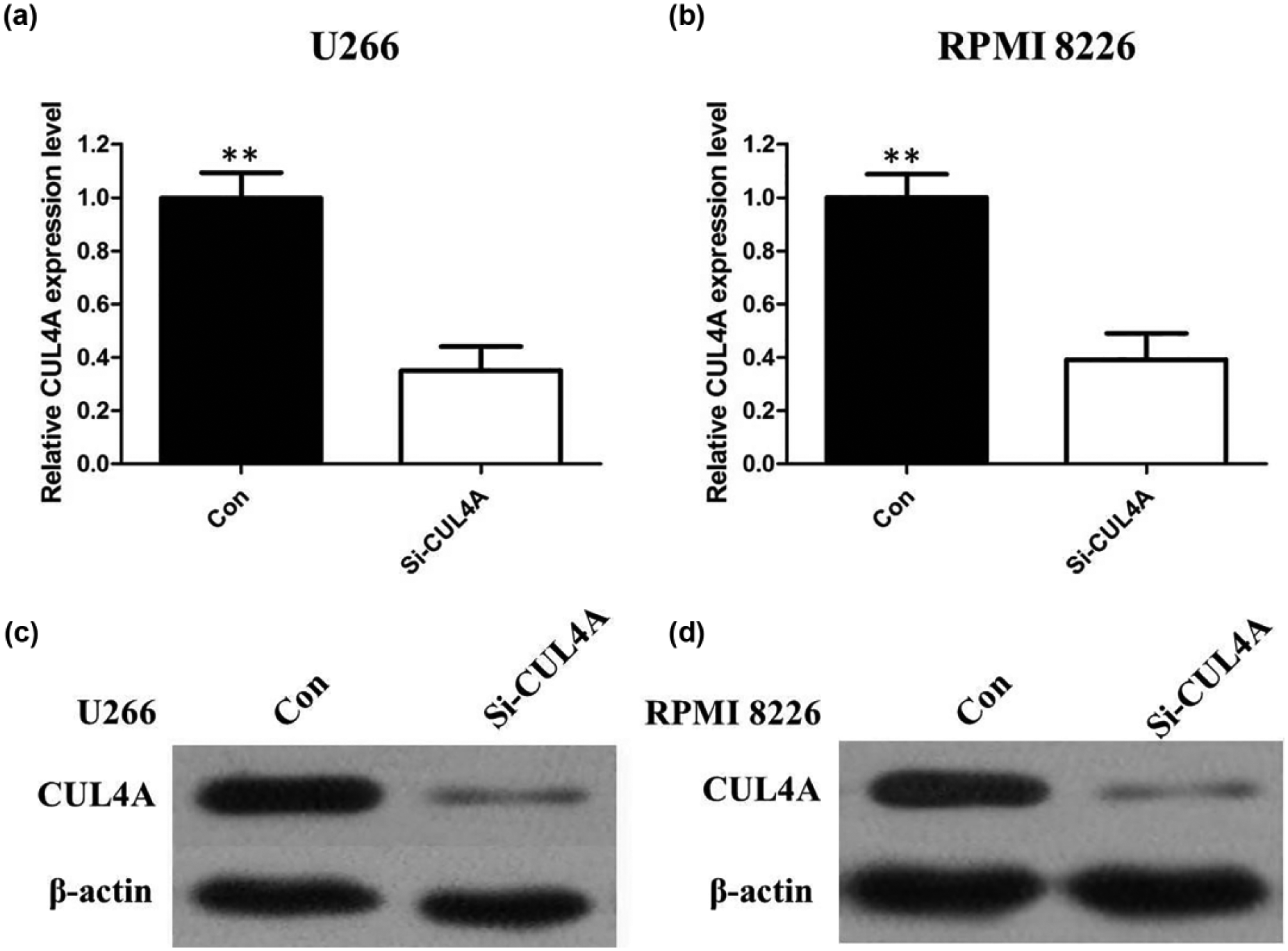
(a) Quantitative real-time PCR shows that siRNA treatment of CUL4A markedly decreased CUL4A mRNA levels in U266 cells. (b) Quantitative real-time PCR shows that siRNA treatment of CUL4A markedly decreased CUL4A mRNA levels in RPMI 8226 cells. (c) Western blots show that siRNA treatment of CUL4A markedly decreased CUL4A protein levels in U266 cells. (d) Western blots show that siRNA treatment of CUL4A markedly decreased CUL4A protein levels in RPMI 8226 cells.

(a) Knocking-down of CUL4A in U266 cells decreased cell proliferation. (b) Knocking-down of CUL4A in RPMI 8226 cells decreased cell proliferation. (c) Colony-formation assay was performed in U266 cells transfected with CUL4A-siRNA. A decrease was seen in the groups with siRNA treatment. (d) Colony-formation assay was performed in RPMI 8226 cells transfected with CUL4A-siRNA. A decrease was seen in the groups with siRNA treatment.
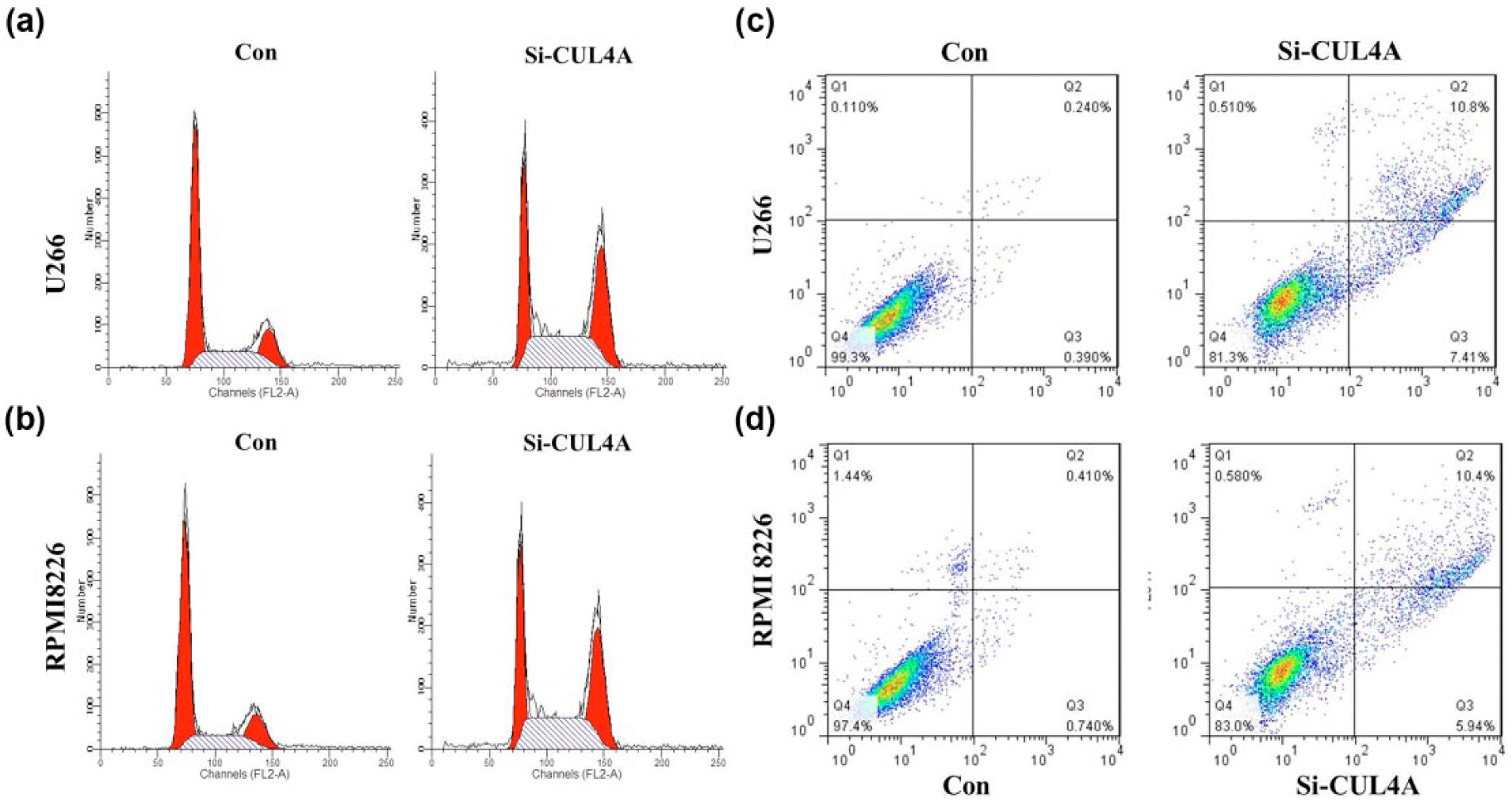
(a) The percentage of cells in G2 and S phases was higher in CUL4A-siRNA U266 than control cells transfected with si-NC. (b) The percentage of cells in G2 and S phases was higher in CUL4A-siRNA RPMI 8226 than control cells transfected with si-NC. (c) Knockdown of CUL4A induced cell apoptosis of U266 cells. (d) Knockdown of CUL4A induced cell apoptosis of RPMI 8226 cells.
CUL4A depletion induces apoptosis of MM cells
Annexin V-FITC analysis was used to detect the rate of apoptosis in U266 and RPMI 8226 cells. The results showed that CUL4A depletion could induce a significant population of early and late apoptotic of U266 and RPMI 8226 cells compared with controls (Figure 4(c) and (d)).
Inhibition of CUL4A suppressed the migration and invasion of MM cells via the epithelial-to-mesenchymal transition pathway
We performed the Transwell assay to determine whether CUL4A was involved in the regulation of migration and invasion of MM cells. Our results showed that knockdown of CUL4A could significantly suppress the migratory and invasion ability of U266 and RPMI 8226 cells (Figure 5(a) and (b); p < 0.01).

(a) Inhibition of migration and invasion of U266 cells by siRNA treatment of CUL4A and (b) inhibition of migration and invasion of RPMI 8226 cells by siRNA treatment of CUL4A.
In order to elucidate the mechanism by which CUL4A regulates MM cell migration and invasion, western blot assays were performed to detect the expression level of epithelial-to-mesenchymal transition (EMT) markers. The results showed that E-cadherin protein expression was upregulated, and protein expression of vimentin and N-cadherin was downregulated in U266 and RPMI 8226 cells transfected with the CUL4A-siRNA (Figure 6(a) and (b)). Therefore, inhibition of CUL4A in MM cells changed the cell morphology from a mesenchymal to a more epithelial phenotype.
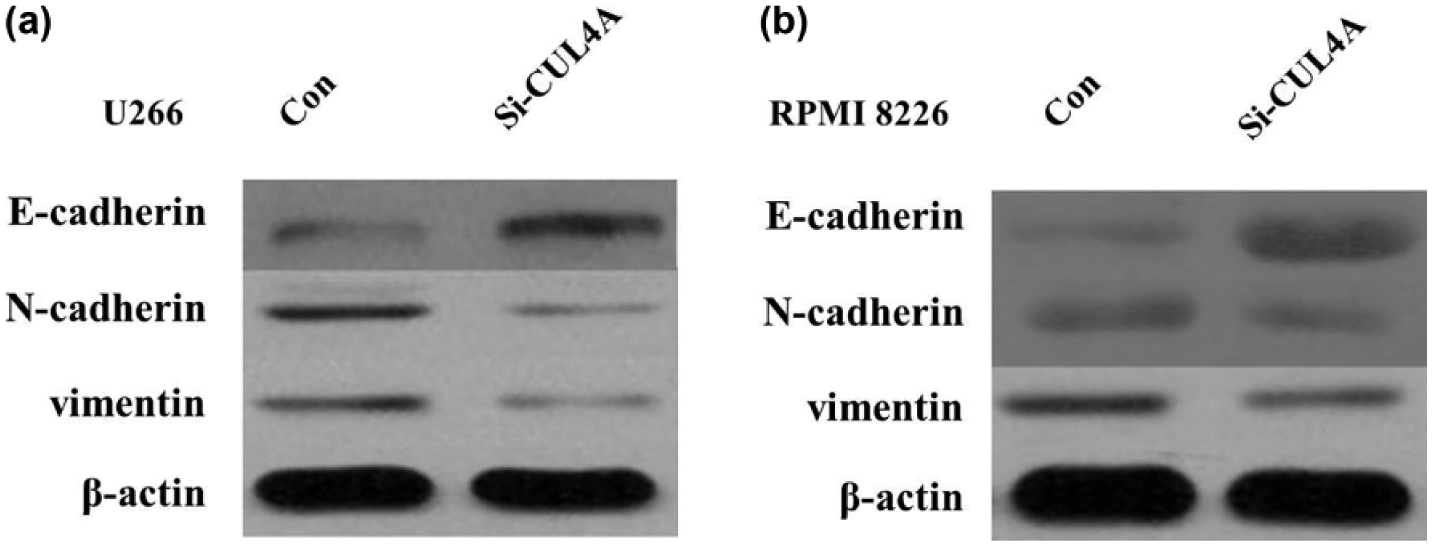
(a) Knockdown of CUL4A reverses EMT in U266 cells and (b) knockdown of CUL4A reverses EMT in RPMI 8226 cells.
Discussion
MM is the second most prevalent blood cancer (10%) after non-Hodgkin’s lymphoma and represents approximately 1% of all cancers and 2% of all deaths from cancer. MM is characterized by the accumulation of clonal serum cells in BM and overproduction of monoclonal immunoglobulin (Ig) in blood or the urine.
1
Since the 1960s, the alkylating drug melphalan (
CUL4A, a member of the cullin family of proteins that composes the multifunctional ubiquitin ligase E3 complex, is essential for the ubiquitination of several well-defined tumor suppressor genes.10,11 Changes in CUL4A potentially exert pleiotropic effects that alter cellular functions. 12 Thus, CUL4A may act as an oncogene, but whether CUL4A plays a role in MM remains unknown. This study explored the potential of CUL4A in MM and investigated whether CUL4A acted as a negative or positive marker in MM. We found that CUL4A expressions were upregulated in MM patients compared with normal controls.
To further illustrate the role and function of CUL4A in MM, we performed the knockdown by small interfering RNA (siRNA) silencing on MM cell lines U266 and RPMI 8226. Our results showed that silencing of CUL4A significantly inhibited cell proliferation, colony formation, migration, and invasion of MM cells. These results demonstrated that CUL4A is associated with carcinoma angiogenesis and malignant transformation and promoted cancer growth of MM, which is consistent with previous study of CUL4A in other cancer cells. Moreover, we further found that knockdown of CUL4A could induce the arrest of G2 and S phases in MM cell lines, which suggests that inhibition of CUL4A may be a potential anticancer agent for patients with MM.
Cell penetration through the Transwell membrane showed increased invasiveness. CUL4A has been reported to be associated with malignant cell behavior in a wide range of human cancers.7,13,14 The evidence suggesting a role of CUL4A in tumor invasion and metastasis has increased. The effect of CUL4A on MM cell invasion was measured by Transwell Matrigel. The results showed that depletion of CUL4A could inhibit cell invasion in vitro. Based on these results, we speculated that invasiveness of MM might be associated with the expressions of CUL4A. To investigate the mechanism underlying the migration process, we measured the protein level of EMT markers following knockdown of CUL4A. EMT is a conserved cellular process in which epithelial tumor cell lacks its polarity and transforms into a mesenchymal phenotype. The feature of EMT occurrence is that the epithelial marker E-cadherin is downregulated and mesenchymal marker, like N-cadherin, is upregulated. Here, we confirmed that knockdown of CUL4A suppressed EMT through the downregulation of vimentin and N-cadherin, consistent with upregulation of E-cadherin in MM cells. Therefore, we propose that CUL4A may be a candidate target for the treatment of MM considering its stimulatory role on angiogenesis, proliferation, cell-cycle phase, and EMT process.
In conclusion, data from this study clearly demonstrate that CUL4A is upregulated in MM samples and cell lines. Inhibition expression of CUL4A induces a marked reduction in MM cell proliferation and an increase in apoptosis. The collective findings improve our understanding of the mechanisms underlying MM progression and support the potential of CUL4A as a promising target for MM diagnosis and therapy.
Footnotes
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.