
Abstract
We previously reported that 37-kDa laminin receptor precursor involved in metastasis of lung adenocarcinoma cancer cells. In this study, we further revealed that hypoxia induced 37-kDa laminin receptor precursor expression and activation of extracellular signal–regulated protein kinase, c-Jun N-terminal kinase, and p38 mitogen-activated protein kinase in lung adenocarcinoma cancer cells. In addition, we further demonstrated that the c-Jun N-terminal kinase inhibitor SP600125 and extracellular signal–regulated protein kinase inhibitor U0126 blocked the c-Jun activity and abolished hypoxia-induced 37-kDa laminin receptor precursor expression and promoter activity in a concentration-dependent manner. However, the p38 mitogen-activated protein kinase inhibitor did not affect 37-kDa laminin receptor precursor expression and c-Jun activity in response to hypoxia. Furthermore, downregulated c-Jun expression by short interfering RNA could also inhibit hypoxia-induced 37-kDa laminin receptor precursor expression and transcriptional activity. The inhibition of 37-kDa laminin receptor precursor expression by SP600125 and U0126 could be rescued by c-Jun overexpression. Studies using luciferase promoter constructs revealed a significant increase in the activity of promoter binding in the cells exposed to hypoxia, which was lost in the cells with mutation of the activator protein 1 binding site. Electrophoresis mobility shift assay and chromatin immunoprecipitation demonstrated a functional activator protein 1 binding site within 37-kDa laminin receptor precursor gene regulatory sequence located at −271 relative to the transcriptional initiation point. Hypoxia-induced invasion of A549 cells was inhibited by the pharmacologic inhibitors of c-Jun N-terminal kinase (SP600125) and extracellular signal–regulated protein kinase (U0126) as well as 37-kDa laminin receptor precursor–specific siRNA or antibody. Our results suggest that hypoxia-elicited c-Jun/activator protein 1 regulates 37-kDa laminin receptor precursor expression, which modulates migration and invasion of lung adenocarcinoma cells.
Introduction
In solid tumors, a hypoxic microenvironment appears to contribute to metastatic progression because the development of metastatic disease is associated with low oxygen levels in clinical studies.1,2 In pancreatic cancer, breast cancer, melanoma xenografts, and lung cancer, hypoxia has been shown to cause hallmarks of malignancy that are associated with metastasis.3,4 In hypoxic cancer cells, the activator protein 1 (AP-1), which supports invasion and metastasis, may be activated.5,6 However, the molecular mechanisms involved in this process are incompletely understood.
Adhesion of cancer cells to endothelial cells is known to be involved in the hematogenous metastasis of cancer under hypoxia.7,8 The 37-kDa laminin receptor precursor (37LRP), also termed MGr1-Ag or p40, is a multifunctional protein. Apart from their potential role as precursors for laminin receptors, numerous studies have linked the 37LRP to tumor metastasis and invasion. The 37LRP is increased in a large variety of cancers in association with their metastatic phenotype.9,10 We recently reported that 37LRP gene expression contributes to metastasis and invasion of adenocarcinoma cancer. 11 The proto-oncogene c-Jun encodes a major component of the AP-1 transcription factor and is a major target of the mitogen-activated protein (MAP) kinase (MAPK) signaling pathways. It consists of dimers of c-Jun/c-Jun, c-Jun/c-Fos, or c-Jun/ATF-2. The AP-1 complex binds to tissue plasminogen activator (TPA) DNA response element (TRE; 5′-TGAG/CTCA-3′), which is present in the promoters of genes that regulate cell proliferation and migration. Among the protein kinases that target c-Jun/AP-1 in vivo, c-Jun N-terminal kinase and extracellular signal–regulated protein kinases 1 and 2 (ERK1/2) are activated. Certain isoforms of p38 MAPKs also are activated by hypoxia. Using BLAST, we predicted a potential binding site in 37LRP promoter region. However, the exact mechanism of the JNK, ERK, and p38 MAPK pathways might regulate 37LRP during hypoxia needs to be investigated.
In this study, we showed JNK, ERK, and p38 MAPK activation and 37LRP protein expression in A549 cells in response to hypoxia in a time-dependent manner. Inhibition of JNK and ERK but not p38 MAPK could block hypoxia-induced 37LRP expression and transcriptional activity. We further elicited c-Jun/AP-1 activity depends on the activation of JNK and ERK under hypoxia. Moreover, we confirm, for the first time, the existence of functional TREs in the 37LRP gene. Finally, we demonstrated that JNK, ERK, and 37LRP are involved in metastasis and invasion under hypoxia.
Materials and methods
Cell culture
The human lung adenocarcinoma cell line A549 was obtained from Shanghai Gene ChemCo Bank (Shanghai, China). The cells were maintained in RPMI 1640 medium containing 10% fetal bovine serum with sodium pyruvate, nonessential amino acids,
Reagents
Rabbit polyclonal antibodies against human total or phosphorylated ERK1/2 (Thr202/Tyr204), stress-activated protein kinase (SAPK)/JNK (Thr183/Tyr185), p38 MAPK (Thr180/Tyr182), and primary monoclonal anti-c-Jun antibody were purchased from Cell Signaling Technology, Inc. (Danvers, MA, USA), and SP600125, SB203580, and U0126 were purchased from Calbiochem (EMD Biosciences Inc., Darmstadt, Germany). The inhibitors were dissolved in dimethyl sulfoxide (DMSO) with the final concentration not exceeding 0.1%. The primary monoclonal anti-37LRP antibody was developed in our laboratory. 12 TransAM AP-1 Kit (Active Motif, Carlsbad, CA, USA) was used for AP-1 transcription factor assay. All other reagents were analytical grade and obtained from the indicated sources.
Western blotting
To examine 37LRP, total or phosphorylated JNK, p38 MAPK, and c-Jun expression, whole cells were harvested and lysed on ice for 30 min in lysis buffer (10 mM Tris (pH 8.0), 1 mM ethylenediaminetetraacetic acid (EDTA), 400 mM NaCl, 10% glycerol, 0.5% NP-40, 5 mM sodium fluoride, 0.1 mM phenylmethylsulfonyl fluoride, and 1 mM dithiothreitol). Equal amounts of protein (25 µg) were loaded onto a sodium dodecyl sulfate polyacrylamide gel (8%–12% polyacrylamide) and subjected to electrophoresis at 200 V for 50 min. Proteins were then transferred to nitrocellulose followed by blocking overnight in blocking buffer (250 mM NaCl, 0.02% Tween 20, 5% goat serum, and 3% bovine serum albumin (BSA)). Primary antibodies were applied in room temperature for 3 h and species-matched peroxidase-conjugated secondary antibodies (1:2000) were then applied for 1 h in room temperature. Labeled bands were visualized by enhanced chemiluminescence (ECL) reagents (Amersham, Piscataway, NJ, USA).
Real-time reverse transcription polymerase chain reaction analysis
Total RNA was extracted from A549 cells exposed to hypoxia at indicated times using RNAzol (Biogenesis, USA) following the manufacturer’s instructions. Synthesis of complementary DNA (cDNA) was performed using a Moloney Murine Leukemia Virus cDNA Synthesis Kit (Gibco BRL, Carlsbad, CA, USA). The cDNAs were then diluted at 1:20 dilution, and 10 µL was used as a template for the TaqMan real-time polymerase chain reaction (PCR) to quantify messenger RNA (mRNA) expression using a qPCR Core Kit (Eurogentec, Belgium) and the ABI Prism 7700 Sequence Detection System (PE Applied Biosystems, USA). TaqMan PCR primers were designed for each gene based on the mRNA sequence using Primer Express Software (PerkinElmer, Waltham, Massachusetts, USA) supplied by Sigma Genosys (USA). The sequences were as follows: 37LRP-F: GCTGGACGATAGCTTGGA and 37LRP-R: GATGACAGATAGCTGGTG; glyceraldehyde 3-phosphate dehydrogenase (GAPDH)-F: ACACTCAGA CCCCCACCACA and GAPDH-R: CATAGGCCCCTC CCCTCTT.
Plasmid constructs and transfection
C-Jun-targeting oligonucleotides for generating cDNA were designed from full-length c-Jun by Shanghai Genechem Co., Ltd (Shanghai, China). Forward: 5′-GGAATTCCACCATGACTGCAAAGATGGAAACGACCT-3′and reverse: 5′-CGGGATCCCGTTATCAAAAT GTTTGCAACTGCTGCGTT-3′. The reverse transcription polymerase chain reaction (RT-PCR) products were then inserted into the vector pcDNA3.1 by EcoRI and BamHI sites (pcDNA3.1/c-Jun). A549 cells were transfected with pcDNA3.1/c-Jun by using Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s protocol. The empty vector was used as a control.
The vector expressing the siRNA of targeting 37LRP and stably transfected subclones were constructed as described in our previous report. 11 The vector expressing the siRNA of targeting c-Jun was constructed as described previously.13,14 In brief, A549 cells were transfected with pSilencer™ 2.1-U6 Neo vector, containing a sequence (5′-CAGCUUCCUGCCUUUGUAA-3′) that specifically targets c-Jun and a scramble sequence (5′-GAUUACU AGCCGUCUUCCU-3′) as control. The sequence was checked against the database to confirm specificity. Transfection was carried out using Lipofectamine 2000 following the manufacturer’s instructions followed by treating the cells with 0.4 mg/mL of G418 to select the transfected cells.
AP-1/c-Jun binding site search
A genomic region of 3000-bp upstream of the 37LRP transcriptional initiation site was determined using the NCBI Genomic BLAST program. This DNA sequence was then pasted into DNA Strider 1.0 software, which was employed to locate putative TRE. The search was based on compilations of functional TRE and the c-Jun/AP-1 binding consensus sequence TGA(C/G) TCA, in turn established by following definitions of consensus sequences. 15
AP-1/c-Jun DNA-binding assay
The nuclear proteins were extracted using a nuclear extract kit following the manufacturer’s instructions. AP-1 binding to the hypoxia response element was assessed using TransAM AP-1 Transcription Factor Assay Kit (Active Motif Europe, Rixensart, Belgium). To accomplish this, an oligonucleotide containing the AP-1 binding site was attached to a 96-well plate. The active form of AP-1 contained in nuclear extracts specifically bound to this oligonucleotide and was revealed by incubation with antibodies using enzyme-linked immunosorbent assay technology with absorbance reading. In total, 10 µµg of nuclear extracts were analyzed for AP-1 binding to the oligonucleotide following the manufacturer’s instructions. The specificity of AP-1 binding was assessed by competition with free wild-type or mutated oligonucleotide following the manufacturer’s instructions.
Transient transfection and luciferase assay
Cells were transiently transfected with reporter constructs (designed as 37LRP-Luc or AP-1-Luc). The AP-1-Luc plasmid was purchased from Clontech (Palo Alto, CA, USA). The construct 37LRP-Luc containing a TRE-like sites or mutated sites from 37LRP promoter corresponding to region from nucleotides −271 to −260 (546 bp containing GCTGACTAACT to GCTTTTTAACT) was synthesized and then inserted into pGL3 promoter vector (Promega, San Luis Obispo, CA, USA). A549 cells in 60 mm dishes were transfected using the Lipofectamine 2000. PRL-TK as a control for transfection efficiency in Dulbecco’s Modified Eagle’s Medium (DMEM) without serum was used; 24 h post transfection, cells were pretreated with or without SB203580 (5 or 50 µM), U0126(1 or 10 µM), or SP600125 (5 or 10 µM) followed by exposing to normixic or hypoxic condition for 24 h. The luciferase activity was measured and quantitated in a luminometer using the Dual-Luciferase Reporter Assay System (Promega). Experiments were performed in triplicates. Results were expressed as mean of the ratio between the firefly luciferase activity and the renilla luciferase activity.
Chromatin immunoprecipitation assay
AP-1 binding to 37LRP promoter was analyzed by chromatin immunoprecipitation (ChIP) assay in the A549 cells as described previously. 7 Briefly, A549 cells were fixed with 1% paraformaldehyde, and chromatin derived from isolated nuclei was sheared with a F550 microtip cell sonicator (Fisher Scientific, MA, USA). After centrifugation, supernatants containing sheared chromatin were incubated with an anti-c-Jun antibody or control IgG. Protein A sepharose was then added, the incubation was continued overnight, and immune complexes were subsequently eluted. Complexes were then treated with RNase and proteinase K and were extracted with phenol/chloroform and then with chloroform. DNA was precipitated, washed, dried, re-suspended in water, and analyzed by PCR. The primers used in this analysis spanned 195 bp around the possibilities of AP-1 binding site located −271 bp from the translation start site (sense, 5′-AGGTACAAGGGCTGGGTAA-3′ and antisense, 5′-CTTCAGGAAGGCGGGCTTG-3′) within 37LRP promoter.
Electrophoresis mobility shift assay
The nuclear extracts were analyzed for AP-1 binding to the 37LRP promoter by gel mobility shift assays (electrophoretic mobility shift assay (EMSA)) as described previously.
16
The DNA probes used in the EMSA experiments contained the following sequences: 5′-GCCTG
A549 cells incubated under hypoxic or normoxic conditions pretreated with or without SB203580 (5 or 50 µM), U0126 (1 or 10 µM), or SP600125 (5 or 10 µM) were harvested and lysed in extraction buffer (20 mM 4-(2- hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES; pH 7.9), 1 mM EDTA, 400 mM NaCl (25%), 0.1% NP-40, 1× protease inhibitor cocktail, 0.5 mM phenylmethylsulfonyl fluoride, and 2.5 mM dithiothreitol). An equal amount of protein (1 µg) from the nuclear extract was used for binding reactions with the biotin-labeled wild-type or mutant 37LRP probe for 20 min at room temperature in binding buffer (8 mM HEPES (pH 7.4), 80 mM KCl, 0.8 mM EDTA, and 1 mM dithiothreitol) at a 20 µL final volume. For super-shift experiments, 1 µg of monoclonal antibody against c-Jun/AP-1 was added to the reaction mixture before the addition of labeled oligonucleotides. Equivalent amounts of biotin-labeled probe were used for all samples. For the binding competition experiment, unlabeled oligonucleotides were added into the reaction mixture in 50- and 200-fold excess. DNA–protein complexes were analyzed in a 4% polyacrylamide gel with 0.5× Tris-borate-EDTA at 100 V. The binding complex was then transferred to nylon membrane at 380 mA for 30 min. Cross-link was carried out for 15 min on a transilluminator equipped with 312 nm. The biotin-labeled DNA was detected by chemiluminescence.
In vitro migration and invasion assays
For Transwell migration assay, 5 × 104 cells in serum-free RPMI 1640 medium were added to the upper chamber of each insert (BD Biosciences, Franklin Lakes, NJ, USA). For invasion assays, the chamber inserts were coated with 50 mg/L Matrigel (BD Biosciences, San Jose, CA, USA)for 4–5 h of incubation at 37°C. The cells (1 × 105 cells) in serum-free RPMI-1640 medium were then added to the upper chamber. In both assays, medium supplemented with serum was used as a chemoattractant in the lower chamber. After incubation in a normoxia (37°C and 5% CO2) or hypoxia (37°C, 1% O2, 5% CO2, and 94% N2) condition for 24 or 48 h, the cells on the upper surface were removed, and the cells on the lower surface of the membrane were fixed with 100% methanol for 15 min, air dried, stained with 0.1% crystal violet, and counted under a microscope (Olympus Corp., Tokyo, Japan). Nine random fields were analyzed per insert. Each experiment was conducted in triplicate in three independent experiments.
Results
Hypoxia activates JNK, ERK, and p38 phosphorylation in vitro and in vivo
MAPKs are activated in response to hypoxia. To explore the signal transduction mechanism involved in 37LRP induction, we first examined whether the known MAPKs, p38 MAPK, ERK, and JNK pathways and 37LRP, were activated by hypoxia. A549 cells were exposed to increasing periods of instantaneous hypoxia by exposing cells in a hypoxia chamber to preconditioned hypoxic medium for 0–4 h. Western blot analysis revealed that hypoxia lead to a rapid and time-dependent increase in JNK, ERK, and p38 MAPK phosphorylation as well as 37LRP expression (Figure 1). These findings revealed that hypoxia induced phosphorylation of JNK, ERK, and p38 MAPK as well as 37LRP protein expression.

Hypoxia-induced phosphorylation of MAPKs and 37LRP expression. Confluent monolayers of A549 cells were exposed to preconditioned hypoxic medium in a hypoxia chamber (1% atmospheric O2) for 0–4 h. Whole cell lysates were separated by SDS-PAGE and immunoblotted for the activated from of JNK, ERK, and p38 MAPK using an anti-phospho-JNK, anti-phospho-ERK, and anti-phospho-p38 MAPK antibody. Data presented were representative of separate experiments.
JNK and ERK inhibition resulted in blockade of hypoxia-induced 37LRP expression and promoter activity
To further explore the MAPK signal pathway in hypoxia-induced 37LRP expression and activity, whole cell lysates from the A549 cells with an upregulation of 37LRP expression after exposure to hypoxia for 8 h were further examined. Pretreatment of the cells with the specific JNK inhibitor SP600125 (5–10 µmol/L) and ERK inhibitor U0126 (1–10 µmol/L) resulted in a significant inhibition of hypoxia-induced expression of 37LRP protein (Figure 2(a)). In contrast, the 37LRP expression was not affected when the A549 cells were pretreated with the specific p38 inhibitor SB203580 (5–50 µmol/L, Figure 2(a)). Similarly, 37LRP mRNA was significantly upregulated in the cells exposed to hypoxia, and SP600125 or U0126 significantly blocked hypoxia-induced 37LRP mRNA upregulation in a concentration-dependent manner. The 37LRP mRNA expression was not affected by SB203580 (Figure 2(b)).

Role of JNK, ERK, and p38 MAPK in mediating hypoxia-induced 37LRP expression. (a) Immunoblotting A549 cells were exposed to hypoxia for 8 h and the cells were harvested with cell lysis buffer. Whole cell lysates were then subjected for immunoblotting as described in the “Materials and methods” section. (b) Quantification of 37LRP mRNA by real-time RT-PCR. A549 cells were exposed to hypoxia for 8 h and total RNA was extracted. Real-time RT-PCR was then performed as described in the “Materials and methods” section (*vs normoxic control, p < 0.05; #vs hypoxia, p < 0.05; ##vs hypoxia, p < 0.01). (c) Luficerase activity of 37LRP promoter. Hypoxia (24 h) increased 37LRP/37LRP promoter activity (N: normoxia; H: hypoxia; *vs normoxic control, p < 0.05). Hypoxia-induced 37LRP promoter activity was inhibited in a dose-dependent manner by JNK inhibitor SP600125 and ERK inhibitor U0126 (#vs hypoxia p < 0.05; ##vs hypoxia p < 0.01). The results shown were representative of three independent experiments.
To investigate whether JNK and ERK has a direct effect on the human 37LRP promoter in A549 cells, luciferase reporter gene assay was then performed to evaluate the effect of p38 MAPK, ERK, and JNK on 37LRP transcriptional activity. The A549 cells transiently transfected with 37LRP promoter (546 bp containing TRE-like sites) linking to a luciferase reporter gene showed an increase in transcriptional activity in response to hypoxia (4.18 ± 0.79-fold, p < 0.05, Figure 2(c)). Pre-incubation of the cells with U0126 (1–10 µmol/L) or SP600125 (5–10 µmol/L) resulted in a significant inhibition of hypoxia-elicited 37LRP promoter activity, while pretreatment of the cells with SB203580 did not alter 37LRP transcriptional activity (Figure 2(c)), indicating that JNK and ERK but not p38 MAPK modulate 37LRP transcription. These findings suggested that hypoxia regulates 37LRP expression and activity through JNK and ERK but not p38 MAPK signaling pathways.
JNK and ERK inhibition resulted in inhibition of c-Jun-dependent transcriptional activity and protein expression
To explore the association between ERK and JNK in c-Jun/AP-1 activation, western blotting, dual-luciferase assay, and c-Jun/AP-1 DNA-binding assay were performed in the presence or absence of SB203580, SP600125, and U0126 in response to hypoxia. The A549 cells transiently transfected with an AP-Luc luciferase reporter plasmid showed a significant induction of luciferase activity in response to hypoxia (5.71 ± 1.21-fold, p < 0.05). SP600125 and U0126 significantly inhibited hypoxia-induced AP-1 DNA-binding and c-Jun phosphorylation, while neither the c-Jun phosphorylation nor the DNA-binding activity was affected by SB203580 (Figure 3(a)–(c))

Role of JNK and ERK in hypoxia-induced c-Jun activity and protein expression. A549 cells were transfected with a luciferase reporter construct under the control of multiple AP-1-Luc before exposure to hypoxia. (a) Effect of JNK inhibitor (SP600125) and ERK inhibitor (U0126) on c-Jun activity. (b) Effect of JNK inhibitor (SP600125) and ERK inhibitor (U0126) on AP-1 activation (*vs normoxic control, p < 0.05; #vs hypoxia p < 0.05; ##vs control under hypoxia, p < 0.01). (c) Effect of JNK inhibitor (SP600125) and ERK inhibitor (U0126) on AP-1 DNA-binding activity. Nuclear extracts were prepared, and AP-1 binding to hypoxia response element oligonucleotide was quantified with TransAM AP-1 transcription factor assay kit. The specificity of binding was assessed by competition with free wild-type (wt) or mutated (mut) oligonucleotide. The results are presented as fold stimulation, which was calculated as the ratio of the different samples to the control (untreated cells; *vs normoxic control, p < 0.05). Hypoxia-induced AP-1 DNA-binding activity was inhibited in a dose-dependent manner by SP600125 and U0126 (#vs hypoxia p < 0.05; ##vs control under hypoxia, p < 0.01). Results shown represent the mean ± SE of three independent experiments performed in duplicate (N: normoxia; H: hypoxia).
Suppression of c-Jun by siRNA resulted in blockade of hypoxia-induced 37LRP transcriptional activity and c-Jun expression modulated 37LRP expression
In order to further explore the relationship between c-Jun/AP-1 function and the hypoxia-induced upregulation of 37LRP, A549 cells were transfected with either a vector containing a scrambled siRNA or a vector containing a c-Jun targeting sequences. After the transfection and antibiotic screening for 6 weeks, the expression of c-Jun in the stably transfected cells was determined by western blotting. The expression of c-Jun in the A549 cells was significantly suppressed by the siRNA (Figure 4(a)). Furthermore, suppression of c-Jun by siRNA significantly reduced expression of 37LRP mRNA (Figure 4(b)) and synthesis of 37LRP protein (Figure 4(c)) in response to hypoxia. Similarly, dual-luciferase assay revealed that inhibition of c-Jun expression blocked hypoxia-induced 37LRP transcriptional activity (Figure 4(d)).

Effect of c-Jun siRNA and sense expression cDNA vector on 37LRP expression and luciferase activity. (a) Effect of c-Jun siRNA on c-Jun expression. After stable transfection of c-Jun siRNA, the expression of c-Jun was evaluated by western blotting. β-actin was used as an internal control. (b) Effect of c-Jun siRNA on 37LRP expression. Western blot analysis was used to evaluate effect of c-Jun on 37LRP expression. (c) Effect of c-Jun siRNA on expression of 37LRP mRNA. Real-time RT-PCR was used for analysis of 37LRP mRNA levels. (d) Dual-luciferase reporter assay was used for analysis of 37LRP promoter activity. (e) After transient transfection of c-Jun sense expression of cDNA vector, the expressions of c-Jun and 37LRP were evaluated by western blotting. β-actin was used as an internal control. (f) Western blot assay suggested that 37LRP expression could be rescued by c-Jun overexpression pretreated with SP600125 (10 µM) or U0126 (10 µM) under hypoxic condition (N: control under normoxia; H: hypoxia; Scr-si: A549 cells transfected scramble sequence; c-Jun/si: A549 cells transfected siRNA targeting c-Jun; c-Jun/cDNA means A549 cells transfected c-Jun sense expression cDNA vector; pcDNA means A549 cells transfected pcDNA 3.1 empty vector). Data presented were representative of separate experiments.
In order to further investigate the effects of c-Jun on 37LRP expression, we constructed c-Jun transient transfection vector and tested the 37LRP expression. Western blot assay suggested that overexpression of c-Jun could promote 37LRP expression (Figure 4(e)). The 37LRP expression will also be detected when c-Jun sense vector and pcDNA vector transiently transfected A549 cell line pretreated with SP600125 and U0126, respectively, under hypoxic condition. The result suggested that the inhibition of 37LRP expression by SP600125 and U0126 could be rescued by c-Jun overexpression (Figure 4(f)).
Confirmation of AP-1 binding sites in 37LRP promoter
Preliminary experiments on luciferase reporter assay demonstrated an increased luciferase activity in the cells transfected with 37LRP promoter. Therefore, we hypothesized that the AP-1 binding site may be located at −423 to +123 from translation start site of the human 37LRP gene. To prove this hypothesis and to assess the contribution of the proximal putative TRE of the 37LRP promoter, luciferase reporter constructs expressing 37LRP promoter were used. As shown in Figure 5(a), hypoxia induced 2.3 ± 0.5-fold increase in luciferase activity in the A549 cells transiently transfected with the wild-type 37LRP promoter (nucleotides −423 to +123) compared to that in the cells exposed to normoxia for 24 h (p < 0.01). However, deletion of the AP-1 binding site at positions −271 to −260 resulted in a nearly complete loss of hypoxia-induced luciferase activity (Figure 5(b), p < 0.01 compared with the wild-type promoter). These findings suggested that this promoter region plays an important role in constitutive 37LRP transcriptional activity.
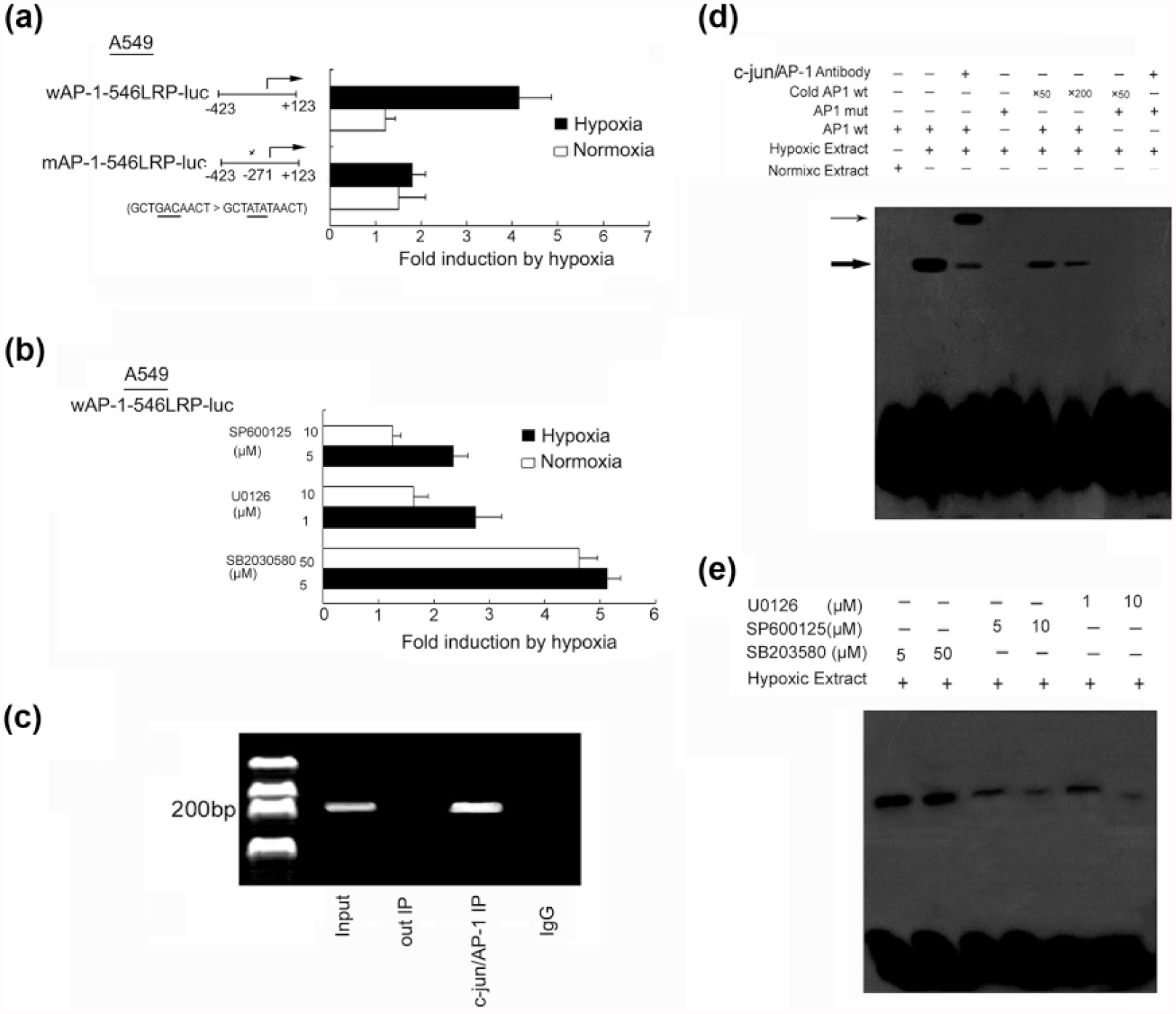
Identification of TRE in the 37LRP promoter and the effect of JNK and ERK on it. Hypoxia induces 37LRP promoter activity in lung adenocarcinoma cancer cells. The predictive consensus binding site at −271 bp were mutated selectively as described in the “Material and methods” section. The mutated sequences of c-Jun/AP-1 were shown. The transcription start site was indicated by an arrow. After transfection with the vector containing consensus binding site at −271 bp or the mutated sequences, the A549 cells were incubated for 24 h in either 21% O2 or 1% O2. A549 cells pretreatment (a) without or (b) with inhibitors SB203580, U0126, and SP600125 incubated for 24 h in either 21% O2 or 1% O2. The luciferase activity was determined, and the results are displayed as fold induction by hypoxia with promoter activity in normoxia normalized to 1. Mean values from three independent transfections were presented (open bars: normoxia; solid bars: hypoxia). (c) Examination of the c-Jun/AP-1 binding to the 37LRP promoter in hypoxic A549 cells by chromatin immunoprecipitation. Reaction control included immunoprecipitation performed using a nonspecific IgG monoclonal antibody (Cntl IP) and PCR performed using whole cell genomic DNA (input). (d) EMSA with a biotin-labeled TRE oligonucleotide was performed as described in the methods. For the competition assay, a 50-fold or 200-fold molar excess of the TRE was used. For the super-shift assay, anti-c-Jun/AP antibody (1 µg) was added to the binding reaction. (e) Effect of JNK, ERK, and P38 on c-Jun 37LRP promoter complex. Specific pharmacologic inhibitors were used. The thick arrow indicates AP-1/DNA complexes, and the thin arrow indicates the super-shift. All results were representative of at least three independent experiments.
As a second approach, the ChIP was performed. As shown in Figure 5(c), ChIP analysis of nuclei derived from A549 cells revealed a dominant band of 216 bp containing the possible binding (−271 to −260) site under hypoxic condition. These results suggested that the proximal TRE is the AP-1/c-Jun binding site in the 37LRP promoter.
EMSA was performed to further confirm aforementioned results. The oligonucleotides corresponding to nt −273 to −251(including AP-1 possibility binding site −271 to −260) of the 37LRP promoter were incubated with nuclear extract derived from the A549 cells exposed to hypoxia for 8 h. The possibility binding site bound strongly to the AP-1 complex (Figure 5(d), lane 2) and also was super-shifted in the presence of anti-AP-1 antibody (Figure 5(d), lane 3). The binding of AP-1 and the 37LRP possibility binding site was prevented by 50-fold and 200-fold excess unlabeled oligonucleotides (Figure 5(d), lanes 5 and 6), indicating the specificity of the interaction. In the absence of AP-1 in the normoxic cell extracts, there is no complex formation on the 37LRP binding site (Figure 5(d), lane 1). To further substantiate the binding of the c-Jun/AP-137LRP complex, the 37LRP possibility consensus site was mutated and found that it was no longer supported complex formation with c-Jun/AP-1 (Figure 5(d), lanes 4, 7, and 8).
Next, we further explored the MAPK signal pathways on AP-1 regulation of 37LRP activity. SP600125 and U0126, but not SB203580, significantly inhibited hypoxia-induced 37LRP promoter activity and binding of c-Jun/AP-1 37LRP complex as demonstrated by dual-luciferase reporter assay and EMSA (Figure 5(b) and (e)). All these findings indicated that the 37LRP promoter is directly activated by AP-1, and the binding site at −271 bp is essential for the transcriptional activation of the 37LRP promoter and ERK and JNK activation.
Suppression of JNK and ERK1/2 and 37LRP inhibited hypoxia-induced metastasis and invasion
In our previous study, we found that 37LRP overexpression contributed to cellular invasion of lung adenocarcinoma cells. To test whether 37LRP is responsible for the increase in cell invasion in response to hypoxia, A549 cells were treated with anti-37LRP antibody and used for cell invasion assay. As shown in Figure 6(a) and Figure 6(b), pretreatment of the cells with anti-37RLP antibody resulted in significant blockade of the cell migration and invasion in response to hypoxia. Consistently, suppression of 37LRP by specific siRNA significantly blocked hypoxia-induced increase in cell migration and invasion. Furthermore, pretreatment of the A549 cells with SP600125 (5–10 µM) or U0126 (1–10 µM) also significantly inhibited hypoxia-induced increase in cell migration and invasion in a dose-dependent manner.

Role of JNK, ERK1/2, and 37LRP on hypoxia-induced cell migration and invasion. (a) Cell migration assay. A549 cells were exposed to hypoxia followed by pretreatment with or without SP600125 (5 or 10 µM) or U0126 (1 or 10 µM), respectively, for 2 h. Cell migration was then assessed as described in the “Materials and methods” section. Hypoxia increased cell migration (*vs cells exposed to normoxia, p < 0.05). SP600125 and U0126 could inhibit the increase in cell migration by hypoxia (*vs cells exposed to hypoxia, p < 0.05). SiRNA targeting 37LRP or preincubation of A549 with 37LRP antibody both decreased cell migration under hypoxia (*vs A549 cells transfected scramble sequence, p < 0.05; *vs A549 cells treated with nonspecific IgG, p < 0.05). (b) Cell invasion assay. A549 cells were pre-incubated with IgG (1 µg/mL) or 37LRP Ab (1 µg/mL), 37LRP siRNA or scramble sequence, SP600125 (5 or 10 µM) then subjected to invasion assay as described in the “Materials and methods” section. Hypoxia increased A549 cell invasive ability (*vs cells exposed to normoxia, p < 0.05). SP600125 and U0126 could inhibit cell invasive ability by hypoxia (*vs cells exposed to hypoxia, p < 0.05). SiRNA targeting 37LRP or preincubation of A549 with 37LRP antibody both decreased cells’ invasive ability under hypoxia (*vs A549 cells transfected scramble sequence, p < 0.05; *vs A549 cells treated with nonspecific IgG, p < 0.05). (c) Cell migration and (d) invasion assay. A549 cells transfected 37LRP expression vector or pcDNA 3.1 empty vector exposed to hypoxia and then pretreated with SP600125 (10 µM) or U0126 (10 µM), respectively, for 2 h. Hypoxia-induced A549 migration and invasion by SP600125 and U0126 could be reversed by 37LRP overexpression (*vs A549 cells transfected empty vector, p < 0.05).
Furthermore, we used the 37LRP overexpression of cell line A549/37LRP previously to perform the migration and invasion assay. 11 The result suggested that the inhibition of hypoxia-induced A549 migration and invasion could be reversed by 37LRP overexpression (Figure 6(c) and (d)).
Discussion
We have previously shown that 37LRP plays a vital role in metastasis and invasion of lung adenocarcinoma cells. 11 However, the role of 37LRP in hypoxia-induced cellular invasion and its underlying molecular mechanisms remain unknown. In this regard, limited data reported that MAPK cascade involved in regulating laminin expression, and the ligand of 37LRP and JNK, ERK1/2, and p38 MAPK play a key role in cell proliferation and invasion in response to hypoxia. 17 This study, therefore, was designed to investigate whether the MAPKs play a role in hypoxia-induced 37LRP expression and subsequent lung cancer cell invasion.
Previous studies by other researchers have shown rapid and transient activation of ERK, JNK, and p38 MAPK in response to hypoxia. 18 In this study, we investigated which MAPK is involved in the signaling cascade of 37LRP induction by hypoxia in a lung adenocarcinoma cell line. Using specific pharmacologic inhibitors, we showed that JNK and ERK activation is necessary for the induction of 37LRP in A549 cells. Pretreatment of cells with SP600125 and U0126 resulted in blockade of the increase in c-Jun/AP-1 expression and activity, the level of 37LRP mRNA, the expression of 37LRP protein, and the transcriptional activity of the 37LRP promoter. However, pretreatment of A549 cells with SB203580, a specific inhibitor of p38 MAPK, did not alter either the induction of c-Jun/AP-1 activity or 37LRP expression and activity in response to hypoxia. These findings suggested that MAPKs, JNK, and ERK signaling pathways mediate induction of 37LRP by hypoxia.
The proto-oncogene c-Jun encodes major component of AP-1 transcription factors, which are important regulators of immediate-early signals directing cellular survival. 19 Among the protein kinases that target c-Jun/AP-1 in vivo, the JNK and ERK1/2 are activated. Certain p38 MAPK also are activated by hypoxia. But these enzymes have not been found to phosphorylate c-Jun. 20 Similarly, we did not observe it on hypoxia-induction of 37LRP expression. We have showed that JNK and ERK1/2 have been activated by hypoxia and subsequently 37LRP expression. We hypothesized that AP-1 might be involved in the regulation of 37LRP by hypoxia through JNK and ERK signaling. Our data revealed significant increase in the level of c-Jun phosphorylation during hypoxia and these increases were dependent on JNK and ERK1/2 activity. Similarly, the inhibition of hypoxia-induced 37LRP upregulation by SP600125 and U0126 could be rescued by c-Jun overexpression. In addition, dual-luciferase assay and DNA-binding assay revealed dramatic increase in AP-1 transactivating potential and their DNA-binding activity exposed to hypoxia, which was also dependent on JNK and ERK1/2 activity. We further investigated whether inhibition of c-Jun by siRNA could block hypoxia-induced 37LRP expression. Inhibition of c-Jun expression could abolish hypoxia-induced 37LRP mRNA level, protein expression of 37LRP, and transcriptional activity. A recent report demonstrated that enhancement of AP-1 binding activity involves transcriptional induction of GRP78 gene in human lung adenocarcinoma cancer cells. In this study, we first reported that JNK and ERK mediated induction of AP-1 activity involved in hypoxia-induced 37LRP gene.
In the case of vascular endothelial growth factor (VEGF), endothelin, and endothelial nitric oxide synthase (eNOS) promoters, cooperation between AP-1 and hypoxia-inducible factor 1 (HIF-1) was observed, 21 and HIF-1 and AP-1 modulate upregulation of CYP3A6 induction by hypoxia. 22 Our previous study found that hypoxia could induce 37LRP expression and further confirmed HIF binding site in the 37LRP promoter in gastric cancer. 23 In contrast, it has also been reported that p53 downregulated 37LRP via AP-2-dependent manner. 24 A search of the cloned gene promoter indicated the possible AP-1 binding sites at 37LRP gene promoter. The dual-luciferase reporter gene assay revealed that hypoxia could induce 37LRP promoter activation, which could be blocked by mutation of the AP-1 possible binding site, suggesting AP-1-dependent induction of 37LRP. Results from the combination of ChIP and EMSA narrowed the sequence −271 to −260 as a classic TRE, and mutation of this AP-1 site resulted in a complete blockade of AP-1 binding activity. Recent studies have also revealed that transforming growth factor-beta 1 (TGF-β1) upregulates AGT transcription in human lung fibroblasts through a mechanism that requires both JunD, a family member of AP-1, and HIF-1α binding to the AGT promoter. 25 However, we did not address HIF-1 and AP-1 collective function in the induction of 37LRP gene in response to hypoxia, and the relationship between HIF-1 and AP-1 on regulation of 37LRP remains to be fully elucidated.
Finally, we examined whether hypoxia-mediated increase in migration and invasion requires signaling through JNK and ERK1/2 activation and subsequent 37LRP upregulation. This study demonstrated that exposure to hypoxia induced a significant increase in 37LRP-mediated migration and invasion of lung adenocarcinoma cancer cells. Pretreatment of A549 cells with the JNK inhibitor SP600125 or ERK1/2 inhibitor U0126 completely inhibited the hypoxia-mediated increase in migration and invasion in a concentration-dependent manner. We also tested whether 37LRP involved in migration and invasion in response to hypoxia. Results revealed that inhibition of 37LRP with siRNA and antibody could both prevent migration and invasion of the cells exposed to hypoxia. Furthermore, we used the 37LRP overexpression of cell line A549/37LRP to perform the migration and invasion assay, and the result suggested that the inhibition of hypoxia-induced A549 migration and invasion by SP600125 and U0126 could be reversed by 37LRP overexpression. These data suggested that hypoxia induced 37LRP expression, which mediates the increase in cell migration and invasion through JNK- and ERK1/2 signaling pathways. It partially explained the mechanism of 37LRP contribution to hypoxia-induced invasion in lung adenocarcinoma cancer.
Taken together, our study shows that hypoxia-c-Jun/AP-1 signaling cascade regulated hypoxia-induced 37LRP expression in lung adenocarcinoma cancer cells. JNK and ERK1/2 signaling contributed to the hypoxia-induced activation of 37LRP, by which mechanism it enhanced A549 cell migration and invasion. These genes are directed toward adaptive responses to hypoxia and prompt survival. Increase in 37LRP expression may contribute to the adaptive responses of the cells to hypoxia and prompt survival and invasion of cancer cells.
Footnotes
Acknowledgements
Y.Z. and Y.W. contributed equally to this work. Y.Z., H.Z, and L.L. conceived and designed the experiments. Yaf.W., Yan.W., and N.Z. performed the experiments. Z.Z. analyzed the data. L.L. and Yaf.W. contributed reagents, materials, and/or analysis tools. L.L. and Yaf.W. wrote the paper. All authors reviewed the manuscript.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Ethical approval
All applicable international, national, and/or institutional guidelines for the care and use of animals were followed. All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the Declaration of Helsinki (1964) and its later amendments or comparable ethical standards.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was funded by the National Natural Science Foundation of China (grant numbers 81572814, 81301990, and 81272348).
Informed consent
Informed consent was obtained from all individual participants included in the study.