
Abstract
Chronic myeloid leukemia is a clonal myeloproliferative disorder that arises from the neoplastic transformation of the hematopoietic stem cell, in which the Wnt/β-catenin signaling pathway has been demonstrated to play an important role in disease progression. However, the role of Wnt signaling antagonists in therapy resistance and disease progression has not been fully investigated. We aimed to study the effects of Wnt/β-catenin pathway antagonists—secreted frizzled-related protein 1 and Wnt inhibitory factor 1—on resistance toward tyrosine kinase inhibitors in chronic myeloid leukemia. Response to tyrosine kinase inhibitors was analyzed in secreted frizzled-related protein 1 and Wnt inhibitory factor 1 stably transfected K562 cells. Experiments were repeated using a tetracycline-inducible expression system, confirming previous results. In addition, response to tyrosine kinase inhibitor treatment was also analyzed using the secreted frizzled-related protein 1 expressing, BCR-ABL positive MEG01 cell line, in the presence and absence of a secreted frizzled-related protein 1 inhibitor. Our data suggests that total cellular β-catenin levels decrease in the presence of secreted frizzled-related protein 1 and Wnt inhibitory factor 1, and a significant increase in cell death after tyrosine kinase inhibitor treatment is observed. On the contrary, when secreted frizzled-related protein 1 is suppressed, total β-catenin levels increase in the cell and the cells become resistant to tyrosine kinase inhibitors. We suggest that Wnt antagonists carry the potential to be exploited in designing new agents and strategies for the advanced and resistant forms of chronic myeloid leukemia.
Keywords
Introduction
Constitutive Wnt/β-catenin signaling is increasingly observed in a variety of malignant tumors.1–6 Wnt/β-catenin signaling has pivotal roles both in early developmental stages and in tissue maintenance of adults. The pathway operates by regulating specific genes that function in cell fate, proliferation, polarity, migration, and differentiation. Signaling leads to the nuclear stabilization of β-catenin, which in turn activates T-cell factor/lymphoid enhancer–binding factor (TCF/LEF)-dependent transcription of its target genes.7,8 Aberrant activation of this pathway has been shown in numerous malignancies. Therefore, mechanisms that may inhibit the abnormal functioning of the pathway are of therapeutic interest. 6 Adenomatous Polyposis Coli (APC) and β-catenin mutations, as well as altered expression of the pathway components represent important mechanisms underlying abnormal activation of Wnt/β-catenin signaling. 9
Deregulated activation of the canonical Wnt pathway is also observed in leukemia.1,10–13 The hematopoietic system of the adult organism is composed of cells with a short life span, which are renewed by differentiating from a small population of hematopoietic stem cells (HSCs). Nuclear β-catenin is a key molecule that drives self-renewal.14,15 The first description of deregulated Wnt signaling in a hematological malignancy was reported in chronic myeloid leukemia (CML). 16 CML is a clonal myeloproliferative disorder that arises from the neoplastic transformation of a pluripotent stem cell and is characterized clinically by an overproduction of granulocytes. The chimeric BCR-ABL gene resulting from the t(9;22)(q34;q11) translocation is considered to be the primary genetic defect in CML, but accumulation of other additional mutations is necessary for disease progression to blast crisis. Normally nuclear accumulation of β-catenin is limited to HSCs but granulocyte–macrophage progenitors from CML patients in blast crisis or therapy resistant disease also display nuclear accumulation of β-catenin and self-renewal capacity. Thus, in the advanced/resistant form of disease, β-catenin and the canonical Wnt signaling pathway are key players of the underlying biology. 16 Tyrosine kinase inhibitors (TKIs) are used as a first-line treatment for CML. Unfortunately, approximately 33% of patients with CML treated with imatinib do not achieve a complete cytogenetic response (CCyR), while others display drug resistance/intolerance to TKIs.17,18 Introduction of novel therapies that address the common mechanisms of resistance in the treatment of CML are needed.
Although the role of Wnt signaling has been well established in the progression and therapy resistance observed in CML, the contribution of/lack of Wnt antagonist proteins has only been suggested but not experimentally investigated. Wnt inhibitor proteins function through the canonical and non-canonical pathways and include members of the “destruction complex” for β-catenin (APC and Axin-2) and antagonists, such as the secreted frizzled-related protein 1 (sFRP1) and Wnt inhibitory factor 1 (WIF1), both which bind directly to Wnt proteins and inhibit their action.19,20 Although mutations of β-catenin, APC, and Axin-2 are frequently observed in a variety of epithelial cancers, they are infrequent in CML and in leukemia in general. 9 Epigenetic silencing of Wnt inhibitors and the stabilization and accumulation of β-catenin in cancers with or without mutational activation of Wnt/β-catenin signaling are another mechanisms that are observed in many cancers.21–25 These proteins are able to inhibit and/or change the activity of Wnt proteins, thereby act as moderators of the signaling cascade and demonstrate a high frequency of aberrant promoter methylation in tumors.
In this study, we aimed to study the role of the secreted Wnt/β-catenin pathway antagonists sFRP1 and WIF1 on resistance toward TKIs in CML, using an in vitro cell line model. We initially showed that the K562 cell line does not express sFRP1 or WIF1, while the MEG01 cell line expresses only sFRP1. Promoter methylation status of sFRP1 and WIF1 genes correlates their expression patterns. We stably transfected sFRP1 and WIF1 expression vectors into the K562 cell line (which does not express either of these genes) and analyzed the response to TKIs by measuring cell death indicators. The same experiments were also repeated using the sFRP1 expressing BCR-ABL positive MEG01 cell line, in the presence and absence of a sFRP1 inhibitor. Our data suggests that when sFRP1 is suppressed, total β-catenin levels increase and the cells become resistant to TKIs. Our results with WIF1 are similar but less striking. On the contrary, total cellular β-catenin levels decrease in the presence of sFRP1 and WIF1 and more pronounced response to TKI treatment is observed.
Materials and methods
Cells lines and reagents
K562 and MEG01 are BCR-ABL positive cell lines derived from CML patients in erythroid and megakaryoblastic blast crisis, respectively. Both cell lines were purchased from Leibniz-Institut DSMZ Deutsche Sammlung von Mikroorganismen und Zellkulturen GmbH, Braunschweig, Germany) and grown in RPMI-1640 medium supplemented with 10% fetal bovine serum (FBS) and 1%
Total RNA extraction and complementary DNA synthesis
Total RNA was extracted from cell lines using RNeasy® Mini Kit (Qiagen, Hilden, Germany). To avoid genomic DNA contamination, RNA samples were treated with RNase-free DNase I (Qiagen) as per the manufacturer’s instructions. RNA purity was verified by OD260/OD280nm absorption ratio. Complementary DNA (cDNA) was synthesized using SuperScript™ First Strand Synthesis System for reverse transcriptase-polymerase chain reaction (RT-PCR; Invitrogen, USA), 2 µg of total RNA, and Oligo(dT)12–18 Primers according to the manufacturer’s instructions.
Relative quantification PCR
Real-time PCR was carried out using a LightCycler® 2.0 (Roche Diagnostics, Mannheim, Germany) instrument and LightCycler FastStart DNA MasterPLUS SYBR Green I Kit (Roche Diagnostics). All reactions were performed in a 20 µL volume with 0.25 pmol of each primer and 5 µL of cDNA template derived from reverse-transcribed RNA. β-actin gene was used as an endogenous control and reference gene for relative quantifications. The same thermal profile was optimized for all primers: a pre-incubation for 10 min at 95°C, followed by 30 amplification cycles of denaturation at 95°C for 30 s, primer annealing at 58°C for 30 s, and primer extension at 72°C for 10 s. H2O was included as a no template control. The analysis step of relative quantification is a fully automated process done using LightCycler Software version 4.0.0.23 (Roche Diagnostics), with the efficiency set at 2. All samples in glass capillaries were later run on 2% agarose gel electrophoresis to check for size and non-specific amplifications. All experiments were done in triplicates.
DNA extraction and sodium bisulfite modification
DNA was recovered from the K562 and MEG01 cell lines using DNeasy Tissue Kit (Qiagen). DNA integrity and purity were verified by gel electrophoresis and OD260/OD280nm absorption ratio, respectively. Sodium bisulfite modification of the DNA was performed using MethylDetector Bisülfite Modification Kit (Active Motif, Europe) according to manufacturer’s instructions.
Bisulfite modification and sequencing
Sodium bisulfite modified DNA template was used for bisulfite sequencing. CpG sites in the promoter region were predicted using MethPrimer 2.0 (PMID:12424112). Sodium bisulfite–treated genomic DNA was used as a template and amplified by sFRP1 primers specific for bisulfite PCR (BS-PCR). Primer sequences are sense primer: 5’-TGGTTTTGTTTTTTAAGGGGTGTTGAGT-3’ and antisense primer: 5′-TCCTACCACAAACTTCCAAAAACCTCC-3′. BS-PCR was carried out in a 25 µL volume using 500 ng of converted DNA as template, 0.4 pmol of each primer, 200 µM of each deoxynucleotide (dNTP), 2.5 mM MgCl2, 2 units of FastStart Taq DNA polymerase (Roche Diagnostics, USA), and 1× of reaction buffer. The thermal profile consisted of a pre-incubation for 5 min at 95°C, followed by 35 amplification cycles of denaturation at 95°C for 45 s, primer annealing at 60°C for 45 s, and primer extension at 72°C for 45 s. H2O was included as a no template control. The PCR products were run on a 4% agarose gel electrophoresis and imaged with INFINITY Bio-1D, Vilber Lourmat (France). All PCR reactions have been independently performed three times. PCR products were subject to bi-directional sequencing (Macrogen, Seoul, Korea).
Cloning and transfection of sFRP1 and WIF1 expression vectors
All primer sequences and restriction enzyme sites used in cloning experiments are given in Supplement 1. NM_003012.4 is the reference sequence for sFRP1. The construction of the sFRP1-pcDNA3.1 expression vector was achieved by initially amplifying the sFRP1 gene by PCR from the HEK293 cell line. The PCR product was ligated using the Kpn1 and BamH1 sites of the vector pcDNA3.1. Transformation of Escherichia coli strain DH5α (New England Biolabs, USA) and selection of transformants were performed as described previously. Transformed cells were plated on Luria-Bertani (LB) agar (Sigma, USA) containing ampicillin (50 µg/mL; Applichem, Darmstadt, Germany). Positive clones were identified by PCR and restriction digestion and grown on LB media. Purification of the expression vectors was accomplished using non-endotoxin plasmid extraction kit (Qiagen). Cells were transfected with Lipofectamine LTX Reagent (3 µL/well; Invitrogen). Stable sFRP1 expressing (designated as K562s) and empty pcDNA3.1 vector control cell lines (designated as K562b) were also generated by growing cells in selective media containing 0.8 mg/mL neomycine.
NM_007191.4 is the reference sequence for WIF1. For the construction of the WIF1 expression vector, the WIF1 gene was amplified by PCR from the HEK293 cell line. Cloning of WIF1 was achieved using pcDNA4/HisMax TOPO TA expression kit (Invitrogen), as per the manufacturer’s instructions. Stable WIF1 expressing cell line (designated as K562w) was generated by growing cells in selective media containing 0.6 mg/mL zeocin. Stable control K562 cell line containing the empty pcDNA4/HisMax vector was also generated (designated as K562wb).
To achieve inducible gene expression of sFRP1, WIF1, and sFRP1 + WIF1, we used a tetracycline-inducible vector system (Tet-Express Inducible expression system; Clontech, USA; Canada). sFRP1 gene was amplified from the HEK293 cell line. Both primers were designed according to the manufacturer’s instructions. The amplified product was cloned into the pTRE3G vector using Sal1 and BamH1 restriction sites as previously described (designated as TET.sFRP1).
For the construction of the WIF1/Tet-On vector (designated as TET.WIF1) the WIF1 gene was amplified from the HEK293 cell line. The amplified product was cloned into the pTRE3G-IRES vector using Sal1 and EagI restriction sites as previously described. Finally, to construct the WIF1/sFRP1 dual Tet-On vector system (designated as TET.WIF1.sFRP1), multiple cloning site I (MCSI) and MCSII of the internal ribosome entry site (IRES), containing pTRE3G vector (Clontech), was used. PCR primers used in the construction of the pTREG-IRES (MCSI)-WIF1 and pTREG-IRES (MCSII)-sFRP1 vectors are also given in Supplement 1. All Tet-On vectors were transiently transfected using Xfect (Clontech) as per the manufacturer’s instructions.
Functionality of the sFRP1 and WIF1 proteins expressed in vectors
Nuclear β-catenin is almost absent in mice control L cells. After treatment with Wnt3a CM, increase in β-catenin levels are observed compared with untreated control cells. Media containing the secreted Wnt antagonists were collected from sFRP1 and WIF1 transfected K562 cells and added to Wnt3a CM treated L cells. In the presence of functional sFRP1 or WIF1 proteins, an inhibition of β-catenin was expected.
Protein extraction and western blotting
Cells were grown to 75% confluency and washed with ice-cold phosphate-buffered saline (PBS). Cell lysis buffer was composed of PYLB buffer supplemented with 50 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES, pH 7.6), 50 mM NaF, 5mM ethylenediaminetetraacetic acid (EDTA), 10 mM sodium pyrophosphate (NaPPi), 1% Triton X-100, 50 mM NaCl, protease, and phosphatase inhibitors (Applichem). Protein content was measured by Bradford assay method (Applichem). β-actin was used as positive control. A volume of 30 µg of each protein was loaded onto 10% sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE), transferred onto polyvinylidene difluoride (PVDF) membranes, and blocked at room temperature for 1 h with Tris-buffered saline containing 0.1% Tween 20 and 5% non-fat dried milk powder. Membranes were blotted with β-actin (1/1000; Cell Signaling Technology, USA), β-catenin (1/1000; BD Biosciences, USA), sFRP1 (1/1000; R&D Systems, Minneapolis)), and WIF1 (1/1000; Santa Cruz Biotechnology) antibodies in PBS containing 0.1% Tween 20 overnight at 4°C. Goat anti-rabbit and goat anti-mouse were used as secondary antibodies (Santa Cruz Biotechnology). Protein bands were detected by Luminata Forte Western HRP Substrate (Millipore, USA) and band intensities were analyzed by ImageJ software (http://imagej.nih.gov/ij/).
Neutralizing sFRP1 in MEG01 cell line by anti-sFRP1 antibodies
Our previous attempts of silencing sFRP1 using siRNAs have only achieved an inhibition of 55.34% (data not shown). We observed that the 55.34% of reduction in sFRP1 expression was not enough to detect any difference in cellular functions. We instead used anti-sFRP1 antibodies neutralizing sFRP1 in MEG01 cell line as an alternative method. The MEG01 cells (4.105/mL) were seeded on six-well plates. A volume of 10 µg sFRP1 antibody (Santa Cruz Biotechnology) was added to MEG01 cells and incubated at 37°C overnight.
Fluorescent microscopy
β-catenin localization and protein levels were analyzed using immunofluorescence labeling. L cells were seeded on coverslips (BD Falcon Culture Slides; BD Biosciences) and treated with Wnt3a CM both alone and together with the addition of CM from sFRP1 transfected cells. All treated cells were incubated and grown at 37°C with 5% CO2 for 6 h. After washing with PBS, the cells were fixed with 4% paraformaldehyde (15 min) and permeabilized in 0.1%–0.2% Tween 20 (15 min) at room temperature, followed by blocking with 1% bovine serum albumin (BSA) for 1 h. Cells were incubated with β-catenin antibody (BD Transduction Laboratories, BD Biosciences, CA, USA) followed by the addition of secondary antibodies (Alexa Fluor 488 goat anti-mouse; Invitrogen). Nuclei were stained by 4′,6-diamidino-2-phenylindole (DAPI; Applichem) and F-actin with Alexa Fluor 568 Phalloidin. Fluorescent microscopy was performed on a Olympus instrument (Olympus BX61 TRF; Olympus, Tokyo, Japan).
Measurement of cell death
Apoptosis was assessed by staining the cells with Alexa® Fluor 488 annexin V and Mitotracker Red. In brief, cells were washed with PBS and suspended in serum-free, phenol red-free medium (in aim to reproduce the same media conditions in which caspase activity was detected). Stable sFRP1 and WIF1 expressing K562 cell lines in addition to TET.sFRP1, TET.WIF1, and TET.WIF1.sFRP1 transiently transfected K562 cell lines and sFRP1 neutralized MEG01 cells were all seeded on six-well plates at a density of 4 × 105 cells/well and treated with IC50 concentrations of TKIs dasatinib, nilotinib, and imatinib for 24 h. After harvesting, cell death was assessed by analyzing mitochondrial membrane potential along with annexin V staining, using MitoTracker® Red and Alexa® Fluor 488 annexin V for flow cytometry kit (Cat. no. V35116; Invitrogen). The cells were subsequently analyzed by a Coulter Epics XL. MCL Flow cytometer (Beckman Coulter Inc. Miami, FL, USA) at 530 to 580 nm wavelength according to the manufacturer’s instructions. Approximately, 10,000 events were collected for each sample. Unstained samples were used as negative fluorescence controls.
Results
K562 cell line does not express sFRP1 or WIF1 while the MEG01 cell line expresses only sFRP1
cDNA derived from the K562 and MEG01 cell lines were used as template in qPCR reactions with the β-actin gene as internal control. Reproducible PCR results show that K562 cells do not express sFRP1 or WIF1, while the MEG01 cell line expresses only sFRP1 (Figure 1(a)). To determine whether promoter methylation was responsible for the repression of sFRP1 expression in K562 cells, methylation status of the sFRP1 promoter was determined by BS-PCR in both K562 and MEG01 cells. BS-PCR revealed that 54 of the 57 CpG sites of the sFRP1 promoter were highly methylated in K562 cells, whereas all CpG sites were unmethylated in MEG01 cells (Figure 1(b)). The sFRP1 promoter methylation pattern correlates with gene expression levels affirming that epigenetic modifications are responsible for sFRP1 gene silencing in K562 cells.

K562 cell line does not express sFRP1 or WIF1 while the MEG01 cell line expresses only sFRP1. Methylation status of the sFRP1 promoter in both the cell lines reveals that epigenetic silencing is responsible for the absence of sFRP1 in K562 cells (a) RT-PCR results showing sFRP1, WIF1, and β-actin gene expression on 2% agarose gel. Hek293 cells were used as control (bands—sFRP1: 435 bp, WIF1:145 bp, β-actin: 202 bp; M-Marker: O’RangeRuler 50 bp DNA ladder; Thermo Scientific Cat. No. SM0613). (b) Methylation status of the CpG islands in sFRP1 gene promoter region of K562 and MEG01 cell lines. Color coding—gray: methylated CpG; red: unmethylated CpG.
sFRP1 and WIF1 transfected cell lines
Stable sFRP1 and WIF1 expressing K562 cell lines have been established by stably transfecting sFRP1/pcDNA3.1 and WIF1/pcDNA4/HisMax expression vectors (along with empty control vectors) into K562 cells. In addition, both the genes were also cloned into a tetracycline-inducible expression system. Tetracycline-inducible transiently transfected cell lines expressing sFRP1 and WIF1 separately and sFRP1 + WIF1 together were generated. Following sequence analyses after cloning, each vector was checked for expression by RT-PCR (Supplement 2).
sFRP1 and WIF1 are functional in transfected cell lines
sFRP1 and WIF1 are both secreted proteins. CM from stably transfected cell lines (and from control stable empty vector–transfected cell lines) was collected and concentrated. Western blotting was performed using sFRP1 and WIF1 antibodies (Figure 2(a) and (b)). We showed that both the proteins were expressed. Nevertheless, the presence of a protein is not necessarily the evidence of functionality. To confirm that the sFRP1 and WIF1 proteins expressed in our stable lines are functional, we used the L cell line which is known to have a very low level of β-catenin expression. 26 Wnt3a is a well-established inducer of β-catenin expression.26,27 Wnt3a CM added to L cells results in an increase in β-catenin expression. When Wnt3a CM is diluted (1:1) with CM from our sFRP1 expressing K562 cell line, a prominent reduction in β-catenin expression is observed both by western blotting and by immune staining (Figure 2(c), Supplement 3). The reduction in β-catenin expression confirms that the Wnt signaling inhibitor sFRP1 expressed in our stable cell lines are functional. Same experimental design was used to evaluate the functionality of the WIF1 protein expressed in our stable cell lines (Figure 2(d)).

(a) sFRP1 protein shown by western blotting in condition media of sFRP1 stably transfected (K562s) and control empty vector–transfected cell lines (sFRP1 antibody; AF1384 anti hsFRP1; R&D Systems, Minneapolis). (b) WIF1 protein shown by western blotting in condition media of WIF1 stably transfected (K562w) and control empty vector–transfected cell lines (WIF1 antibody; SC-30791; Santa Cruz Biotechnology, Santa Cruz, CA, USA). (c) sFRP1 secreted into the CM of our stable cell lines is functional. An increase in β-catenin levels are observed in Wnt3a CM treated L cells, when compared with control. This increase is repressed when cells are co-treated with sFRP1 CM. The primary antibodies used are the following: β-actin; Cell Signaling Technology; β-catenin; Cat. No. 610153; BD Transduction Laboratories (d) WIF1 secreted into the CM is functional. β-catenin levels are suppressed when co-treated with WIF1 stable cell line CM.
β-catenin levels are reduced in transfected cell lines
β-catenin levels in stable sFRP1 and WIF1 transfectants were observed to be reduced when compared to control empty vector stable transfectants in K562 cells (Figure 3(a)). β-catenin levels were calculated by the densitometric analysis of bands following western blotting and normalization of β-actin levels. The same experimental design was employed to tetracycline-inducible vectors TET.sFRP1, TET.WIF1, and TET.WIF1.sFRP1. Half of the transfected cells were treated with the transactivator after which β-catenin levels were compared. In line with previous observations, β-catenin protein levels were reduced in K562 cells expressing Wnt signaling inhibitors sFRP1 and WIF1 (Figure 3(b)).

β-catenin levels are reduced in K562 cells transfected with Wnt signaling inhibitors sFRP1 and WIF1. (a) Western blotting and densitometric analyses of the total change in β-catenin levels in K562s, K562b, K562w, and K562wb cells. (b) Western blotting and densitometric analyses of the total change in β-catenin levels in transactivator treated and untreated Tet-On vector system experiments. (c) MEG01 cells express sFRP1. An opposite approach using an antibody to neutralize sFRP1 was performed. An increase in β-catenin expression was observed after antibody treatment.
We used an opposite approach in MEG01 cells that express sFRP1. We treated MEG01 with a sFRP1 antibody to neutralize the sFRP1 protein. We showed that β-catenin levels increased in the presence of Wnt3A in the neutralized cells, as expected (Figure 3(c)).
sFRP1 and WIF1 expression increases cell death in TKI treated K562 cells
sFRP1 and WIF1 stably transfected K562 cells and tetracycline-inducible TET.WIF1, TET.sFRP1, and TET.WIF1.sFRP1 transiently transfected K562 cells were treated with previously determined IC50 concentrations of TKIs—0.5 µM imatinib, 0.01 µM dasatinib, and 0.1 µM nilotinib—for 24 h, after which the cell death was determined by analyzing mitochondrial membrane potential along with annexin V staining. The data presented are the cumulative result of triplicate experiments repeated independently three separate times (Figure 4).

Cell death levels (normalized) observed after treatment with TKIs in K562s/K562b, TET.sFRP1 (TA+)/TET.sFRP1 (TA−) cells, K562w/K562wb cells, TET.WIF1 (TA+)/TET.WIF1 (TA−) cells, TET.WIF1.sFRP1 (TA+)/TET.WIF1.sFRP1 (TA−) cells, and MEG01 cells, after neutralization with sFRP1 antibody/normal MEG01 cells (TKI, tyrosine kinase inhibitor; K562s, K562/sFRP1/pcDNA3.1; K562b, K562/pcDNA3.1; TET.sFRP1 (TA−), transactivator negative; TET.sFRP1 (TA+), transactivator positive; K562w, K562/WIF1/pcDNA4; K562wb, K562/pcDNA4; TET.WIF1 (TA−), transactivator negative; Tet.WIF1 (TA+), transactivator positive; and TET.WIF1.sFRP1 (TA−), transactivator negative; TET.WIF1.sFRP1 (TA+), transactivator positive).
K562 cells stably expressing sFRP1 were found to be 75%, 43%, and 48% more sensitive to imatinib, dasatinib, and nilotinib, respectively, when compared to empty vector–transfected controls. In TET.sFRP1 cells, a 43%, 17%, and 7% increase in cell death was observed in imatinib, dasatinib, and nilotinib treated cells respectively, in the presence of the transactivator (Figure 4).
K562 cells stably expressing WIF1 were found to be 8%, 13%, and 0.96% more sensitive to imatinib, dasatinib, and nilotinib, respectively, when compared to empty vector–transfected controls. In TET.WIF1 cells, a 59%, 21%, and 2% increase in cell death was observed in imatinib, dasatinib, and nilotinib treated cells, respectively, in the presence of the transactivator (Figure 4). In TET.WIF1.sFRP1 cells a 27%, 19%, and 10% increase in cell death was observed after imatinib, dasatinib, and nilotinib treatment, respectively, in presence of the transactivator (Figure 4).
In K562 cell transiently transfected with WIF1/pTRE3G-IRES (TET.WIF1), apoptotic response after imatinib treatment was assessed by western blotting using a poly (ADP-ribose) polymerase (PARP) antibody. TET.WIF1 cells treated with transactivator + imatinib revealed a higher degree of PARP cleavage compared to cells treated with transactivator only, suggesting that increased WIF1 expression renders cells more vulnerable to TKIs (Figure 5).
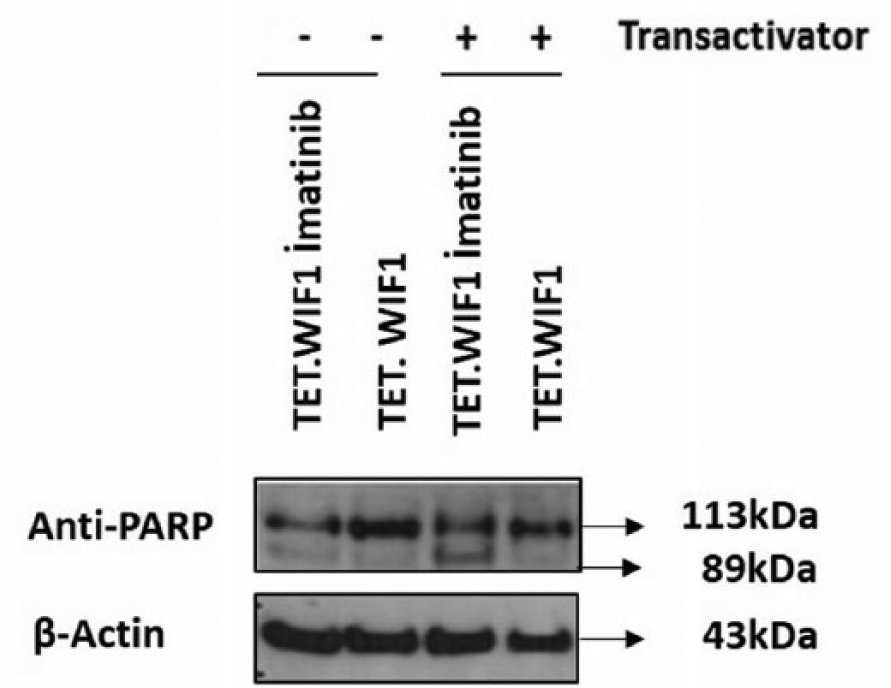
WIF1/pTRE3G-IRES transfected K562 cells treated with 0.5 µM imatinib for 24 h. Western blotting performed with anti-PARP antibody reveals PARP cleavage after imatinib treatment. Image shows two biological replicas.
When previous data on β-catenin levels in our transfected cell lines and cell death response to TKIs are combined, we concluded that a reduction in β-catenin levels corresponds to increased sensitivity toward TKIs.
sFRP1 silencing leads to resistance in TKI treated MEG01 cells
In MEG01 cell that expresses sFRP1, we used an experimental design in which we neutralized sFRP1 activity by specific antibodies. We observed that neutralized cells gained resistance to TKI treatment. A 48%, 48%, and 51% resistance to cell death was observed in cells treated with imatinib, dasatinib, and nilotinib, respectively, when compared to control cells. In other words, increase in β-catenin levels corresponds to resistance to TKI treatment (Figure 4).
Discussion
Wnt antagonists/inhibitors function as tumor suppressors. Silencing of these genes via hypermethylation of their promoters has been shown to be part of the pathogenesis in a plethora of human malignancies.1–6 Aberrant regulation of the Wnt signaling pathway is known to play a role in tumorigenesis and maintenance of cancer stem cells.3,4,28 The Wnt pathway can be activated aberrantly not only by mutations of genes functioning in Wnt signaling such as APC and CTNNB1 but also by dysregulation of secreted antagonists. For example, diminished expression caused by promoter methylation of Wnt antagonists genes sFRP1 and WIF1 results in unregulated increase of ligand-dependent Wnt signaling activity. Although the aberrant expression of sFRP1 and WIF1 as Wnt inhibitors for both canonical and non-canonical pathways was reported to be involved in the tumorigenesis of different kinds of cancers, including leukemia, association of their protein expression patterns with TKI resistance observed in CML remains unclear. This study provides the first evidence that induced sFRP1 and WIF1 expression will sensitize CML cells to TKIs.
We showed that K562, a blastic phase CML cell line, does not express sFRP1 due to promoter hypermethylation. Forced expression of sFRP1s in these cells sensitizes them to TKI treatment. We also showed that forced WIF1 expression and sFRP1 + WIF1 expression together have similar outcomes of increasing the effects of TKI administration. However, in control experiments in which the sFRP1 protein is neutralized in MEG01 cells (MEG01 cell line normally expresses sFRP1), a gained resistance to TKI treatment was observed. To summarize, the restoration of sFRP1 expression in K562 cells by ectopic expression inhibits Wnt activity accompanied with destabilization of β-catenin. Importantly, restoring sFRP1 levels induced apoptotic cell death. However, inhibiting sFRP1 expression in MEG01 cells stabilizes β-catenin and results in higher degree of resistance toward TKIs. This study supports the critical role for sFRP1 and WIF1 silencing in CML and reinforces the importance of the Wnt antagonists in preventing oncogenic stabilization of β-catenin and chronic activation of the canonical Wnt pathway, suggesting that sFRP1 and WIF 1 may be an attractive target for an early cancer detection and therapeutic intervention. We previously reported that the combined effects of imatinib and a demethylating agent increased cell death 2.5-fold in K562 cells, suggesting epigenetic modulations such as sFRP1 promoter methylation may interfere with imatinib efficacy. 29
Nuclear accumulation of β-catenin is limited to HSCs, where it is the driving force of self-renewal.8–11 Granulocyte–macrophage progenitors from CML blast crisis or therapy resistant disease also displays self-renewal capacity, a feature they normally do not posses. 16 Aberrant Wnt signaling and β-catenin nuclear accumulation in granulocyte–macrophage progenitors were shown to be responsible for this observation. Our group and others have reported that β-Catenin activation through mutations does not occur in CML.9,30,31 As a result, the underlying mechanisms of activated Wnt signaling have not been identified. One probable mechanism would be the suppression of Wnt antagonists such as sFRP1. DNA methylation of CpG sites in the promoter region has been demonstrated to be responsible for the downregulation of sFRP genes in many forms of cancer and leukemia.21,32–36 Our data is in line with previous studies, showing that transcriptional silencing of sFRP1 correlates with therapy resistance and progressive disease. 29 We suggest a model of hematopoiesis in which Wnt antagonists sFRP1 and WIF1 normally play a central role in downregulating β-catenin in granulocyte–macrophage progenitors. CML progression correlates with the epigenetic silencing of these genes and results in the nuclear accumulation of β-catenin in granulocyte–macrophage progenitors, which in turn leads to the gain of self-renewal function.
Activated Wnt signaling may be a key molecular event, which is able to circumvent the inhibitory effects that imatinib has on BCR-ABL signaling, whether it be in disease progression or primary resistance to therapy. We have shown for the first time that Wnt antagonist expression sensitizes CML cells to TKIs, suggesting that Wnt antagonists may carry the potential to be exploited in designing new agents and strategies for the advanced and resistant forms of CML.
Conclusion
We suggest that Wnt antagonists carry the potential to be exploited in designing new agents and strategies for the advanced and resistant forms of CML.
Footnotes
Acknowledgements
We would like to thank H.O. and K.K. for scientific discussions and H.A. for his assistance in fluorescence-activated cell sorting (FACS) analyses. All authors approved the final version of the manuscript for submission.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported, in part, by The Scientific and Technological Research Council of Turkey (TUBITAK; Research Grant No. 111S480). M.P. performed the research, analyzed the data, and drafted and wrote the manuscript; C.C. performed research; Z.Y. analyzed and interpreted the data and drafted and wrote manuscript; H.O.S. designed the research, analysis, interpreted the data, and drafted the manuscript.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.