
Abstract
Fibrinogen Asn-Gly-Arg motif can specifically recognize and bind to Aminopeptidase N (CD13) on vascular endothelial cells in newly formed tumor vessels. Adipose-derived stem cells can serve as ideal vectors for gene therapy because of their ability of migrating to tumor tissues. First, this study was aimed to design a new peptide (CNGRCLLII(KLAKLAK)2) named CNAK which contains cyclic Asn-Gly-Arg motif and test its biological activity against human umbilical vein endothelial cells. Second, we aimed to construct stably transfected adipose-derived stem cells which express the CNAK peptide and investigate their anti-angiogenic activity in vivo. Adipose-derived stem cells were employed to localize CNAK on vascular endothelial cells in tumors based on their homing property. First of all, the new peptide was synthesized, which effectively entered into CD13+ human umbilical vein endothelial cells and showed cytotoxicity against human umbilical vein endothelial cells. The peptide induced apoptosis of human umbilical vein endothelial cells in a time- and dose-dependent manner, inhibited the expression of Bcl-2, and promoted the expression of Caspase-3 in human umbilical vein endothelial cells. Furthermore, the migration and tube formation of human umbilical vein endothelial cells were inhibited by CNAK. Primary adipose-derived stem cells were then isolated and identified. Stably transfected adipose-derived stem cells which express CNAK peptide (CNAK-ASCs) were successfully established, and the migration of CNAK-ASCs was assessed. In vivo, CNAK-ASCs were found to inhibit the growth and angiogenesis of breast cancer xenografts. This effect may be through inhibiting the secretion of matrix metalloproteinase-2 and membrane type 1-matrix metalloproteinase in vivo. It was also found that CNAK-ASCs reduced the quantity of breast cancer stem cells in tumor tissues. Our data suggested that the new peptide CNAK containing Asn-Gly-Arg motif had anti-angiogenic activity in vitro and in vivo.
Introduction
Breast cancer is one of the most common cancers of female patients. Although the early diagnosis and comprehensive treatment of breast cancer has been significantly improved, the death rate is still unsatisfied on account of tumor metastasis, relapse, and treatment resistance. Folkman et al. 1 in 1971 first identified the relationship between tumor growth and angiogenesis. The size of the tumor will not exceed 2–3 mm if without blood vessels. Therefore, angiogenesis plays an important role in tumor progress. 2
Adipose stem cells (ASCs), isolated from adipose tissues, have many biological characteristics similar to those of bone marrow mesenchymal stem cells (BSCs).3,4 A variety of studies have revealed that ASCs possess an active homing potential for multiple types of cancer.5–8 Besides, ASCs are easily obtained, cultured, and transfected in vitro and can survive after transplantation into animals.9–11 Based on these, ASCs are promising candidates for targeted delivery of anti-angiogenesis drugs to the microvessels of tumor tissues.
Asn-Gly-Arg (NGR) motif, a repetitive structure of fibronectin, can specifically recognize and bind to CD13 receptors (Aminopeptidase N) on vascular endothelial cells of newly formed tumor vessels and tumor cells. 12 NGR motif also can undergo deamination of asparagine to form an isomer, isoDGR. The receptor of isoDGR integrin αυβ3 which belongs to integrin family is overexpressed in newly formed tumor vessels and tumor cells and can meditate adhesion and migration of endothelial cells.13,14 Based on these, NGR peptide has been used for targeted delivery of various antitumor compounds because it can deliver drugs to CD13+ tumor neovascular endothelial cells and tumor cells. 15 Cell-penetrating peptides with 5–30 amino acids can assist drugs to enter cells and improve the antitumor efficacy because of their permeability of cellular membrane. Thus, they have been considered as promising therapeutic agents for the intervention of cancer. 16 However, whether a cell-penetrating peptide containing the NGR motif can modulate anti-angiogenesis activity and how this NGR-bearing peptide affect the growth of endothelial cells in vitro and in vivo have not been systemically explored.
In this study, a new peptide (CNAK) containing the cyclic NGR motif was designed, and its biological activity against human umbilical vein endothelial cells (HUVECs) was studied. Stably transfected ASCs which express CNAK peptide (CNAK-ASCs) were established, and their anti-angiogenic property in vivo was investigated. ASCs were used to localize CNAK on vascular endothelial cells in tumors based on their homing property.
Materials and methods
Synthesis of the peptides
The sequence of the peptides is shown in the following:
CNGRCLLII(KLAKLAK)2 (CNAK);
CNGRCLLIIKKALAAKLLKKALK (CNRK).
Cys-Asn-Gly-Arg-Cys (CNGRC) is a classic motif of fibronectin, which specifically binds to CD13. Leu-Leu-Ile-Ile (LLII) is a linker, connecting two functional domains. The K residue at C terminal of the amino acid was marked with Rhodamine B. Lys-Leu-Ala (KLA) is derived from antimicrobial peptides which is rich in amphiphilic amino acids. It can damage mitochondrial membrane structure and result in apoptosis and necrosis. CNRK was a random sequence of CNAK. Both CNAK and CNRK were synthesized and purified by high-performance liquid chromatography (HPLC) and mass spectrometer (MS; Terabio Biotechnology Company, Guangzhou, China).
Cell lines
Human breast cancer cell lines MCF-7 and MDA-MB-231 were purchased from Typical Cell Culture Collection Committee of the Chinese Academy of Sciences Library (Shanghai, China). HUVECs, human renal tubular epithelial cell line HK-2, and mouse fibroblast cell line L929 were obtained from American Type Culture Collection (ATCC, Manassas, VA, USA). Adipose-derived stem cells (ASCs) were cultured in Dulbecco’s modified Eagle’s medium (DMEM)/F12 medium containing 10% fetal bovine serum (FBS; HyClone Laboratories, Inc., Logan, UT, USA), 100 U/mL penicillin, and 100 U/mL streptomycin (Invitrogen, Shanghai, China). Other cells were cultured in DMEM or Roswell Park Memorial Institute (RPMI) 1640 medium supplemented with 10% FBS, 100 U/mL penicillin, and 100 U/mL streptomycin. All cells were incubated at 37°C in an incubator with 5% CO2 and saturated humidity.
Cell proliferation assay
Cytotoxicity of CNAK for HUVECs was assessed by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay. HUVECs were seeded in 96-well plates (Corning Inc., Corning, NY, USA) at a density of 5 × 104 cells per well. Different concentrations of CNAK and CNRK were added in the wells (ranging from 25 to 200 µg/mL) for 24, 48, and 72 h at 37°C. Then, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT; Sigma, St. Louis, MO, USA) solution (5 mg/mL) was added into each well and incubated for additional 4 h at 37°C. Then, the medium was removed, and 100 µL of dimethyl sulfoxide (DMSO; Sigma) was added to each well to dissolve the formazan crystals, and the optical density (OD) value at 570 nm was measured using a microplate reader. Wells without cells were used for the blank of the spectrophotometer.
To assess the proliferation of ASCs, the exponentially growing third-passage ASCs were (2 × 103 cells/well) plated into 96-well plates and incubated at 37°C. 10 µL MTT solution (5 mg/mL) was added into each well and incubated for additional 4 h at 37°C. The supernatant was removed and 100 µL of DMSO was added, followed by shaking for 10 min to dissolve the crystals. The absorbance was measured by a microplate reader at 570 nm. With time as the horizontal axis, with OD value as the vertical axis, growth curve was drawn. The proliferation of HUVECs and ASCs was determined using the GraphPad Prism (Version 5.02; GraphPad Software, La Jolla, CA, USA).
Flow cytometry
Annexin V/propidium iodide (PI) double notation was used to detect the apoptosis rate of HUVECs. Cells were washed two times with phosphate-buffered saline (PBS) and then harvested with 0.05% trypsin after being treated with CNAK, CNRK, and control (normal media) for 24 h. Following centrifugation, cells were then resuspended in 500 µL binding buffer and 10 µL Annexin V/FITC (20 µg/mL) and 5 µL PI (50 µg/mL) for 5 min at room temperature. The samples were then analyzed by flow cytometry on a FACSCalibur flow cytometer (BD Biosciences, San Diego, CA, USA).
Surface markers CD29, CD90, CD31, CD45, and CD106 were analyzed to identify ASCs. The third-passage ASCs were harvested with 0.05% trypsin. Detached cells were washed three times with PBS, centrifuged (1200×g) for 10 min, and resuspended in the 100 µL PBS (104 cells/100 µL). Nonspecific antigens were blocked by 1 µL 5% bovine serum albumin (BSA). Fluorochrome-conjugated monoclonal antibodies PE-CD29 2 µL, PE-CD90 0.5 µL, PE-CD45 0.5 µL, PE-CD106 5 µL, and fluorescein isothiocyanate (FITC)-CD31 0.5 µL (eBioscience, Santiago, CA, USA) or their respective isotype controls were added to the cell suspension at concentrations recommended by the manufacturer and incubated at 37°C in the dark for 30 min. The labeled cells were washed in the PBS and centrifuged (1200×g) for 10 min. The supernatant was removed and the cells were resuspended in 500 µL PBS. Then, the cells were determined by flow cytometry on a FACSCalibur flow cytometer.
Cellular uptake study
Cells were seeded on 24-well plates and treated with 20 µg/mL CNAK. Following 12 and 24 h incubation, the cells were washed three times with PBS and fixed with 4% formaldehyde for 10 min and permeabilized in PBS with 1% Triton X-100 for about 10 min. After washing with phosphate-buffered saline with Tween 20 (PBST), cells were stained with 4′,6-diamidino-2-phenylindole (DAPI; diluted in 1 µg/mL by PBS) for 10–15 min. Fluorescence images were captured at 200× magnification under a fluorescence microscope (ECLIPSE Ti; Nikon, Tokyo, Japan). In this study, MDA-MB-231 cells and HK-2 cells were also tested for 24 h.
Western blot analysis
Cells were treated with CNAK (100 µg/mL), CNRK (100 µg/mL), and control (normal media) for 24 h. After washed with cooled PBS three times, the cells were treated with 200 µL radioimmunoprecipitation assay (RIPA) Lysis (Pioneer Technology, Xi’an, China) with protease inhibitors (2 mg/mL) for 30 min. Then, the proteins were subjected to 12,000 r/min at 4°C for 20 min. Protein concentrations were measured and cell extracts were then separated by 10% sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and transferred onto polyvinylidene fluoride (PVDF) membranes (Millipore, Billerica, MA, USA). The membranes were incubated with rabbit anti-human Caspase-3 (Abcam, Cambridge, UK), rabbit anti-human Bcl-2 (Abcam), and rabbit anti-human CD13 (Abcam) overnight at 4°C. Then, the proteins were incubated with the corresponding secondary antibodies (Santa Cruz Biotechnology Inc., Santa Cruz, CA, USA) for 1 h. The blots were detected by enhanced chemiluminescence (ECL; Thermo Fisher Scientific, Rockford, IL, USA) on X-ray film. Image-Lab Software was employed to measure the expression of proteins (Bio-Rad, Hercules, CA, USA).
After infection, ASCs infected with CNAK were screened by puromycin (Sigma). Then, the expression of CNAK was determined by western blot. The steps were the same as mentioned above, and the first antibody was mouse anti-flag tag monoclonal antibody (1:1000; Abcam).
Transwell assay
Transwell assay was used to determine the migration ability of HUVECs (Transwell Chamber; Millipore). Cells were resuspended at a density of 1 × 105 cells/mL in 200 µL serum-free RPMI 1640 media containing CNAK or CNRK (20 µg/mL) and seeded in the upper chamber. The lower chamber was filled with 600 µL RPMI 1640 medium containing 10% FBS. After incubation for 24 h at 37°C in 5% CO2, the unmigrated cells were erased using a cotton swab. The migrated cells on the filter membrane were fixed in 4% paraformaldehyde (PFA) for 30 min and stained for 20 min in crystal violet. Then, the cells were visualized and counted in each of five random sights using an inverted phase-contrast microscope at 200× magnification (TMS; Nikon).
The effect of gene transfer on migration of ASCs was assessed by Transwell assays. The migration assay was performed as previously described. ASCs of 200–300 µL with density of 1 × 105 cells/mL were seeded in the upper chamber in a serum-free DMEM/F12 medium. The lower chamber was MDA-MB-231-conditioned medium (MM231-CM), mouse fibroblast cells L929-conditioned medium (L929-CM), and normal medium containing 10% FBS. The rest of the procedure was the same as above.
Wound healing of HUVECs
After trypsinization, HUVECs (5 × 104 cells/mL) were seeded in six-well plates. Following cell fusion to 50%–60%, the cells were scraped by a 100-µL micropipette tip. The cells were treated with 3 mL serum-free medium containing CNAK or CNRK (20 µg/mL). Every12 h incubation, cells were captured and observed and the average distance of the wound was measured. The relative motility of HUVECs was in accordance with the following formula: (the distance between cells at 0 h − the distance between cells at 36 h/72 h)/the distance between cells at 0 h × 100%.
Tube formation assay of HUVECs
The Matrigel (BD Biosciences) was used to test the tube formation ability of HUVECs affected by CNAK. The Matrigel (50 µL) was planted to 96-well plates and placed for 30 min at 37°C for solidification. HUVECs (2 × 105) with pretreatment of CNAK (20 µg/mL), CNRK (20 µg/mL), or normal media were trypsinized, resuspended in 100 µL of MDA-MB-231-CM, and injected in the solidified Matrigel. The cells were observed every 4–6 h using an inverted phase-contrast microscope, and five fields which were randomly selected were captured at 100× magnification. The tubes formed by HUVECs were counted using Image-Pro Plus Software.
Isolation and culture of primary ASCs
Adipose tissues were extracted and isolated from bilateral inguinal region of adult male SD rats. The tissues were washed two to three times by PBS to remove fascia and vessels. Then, the tissues were minced into pieces (0.8–13 mm) using a sterile surgical blade followed by enzymatic digestion of 0.1% collagenase I for 1 h at room temperature. After agitation (120 r/min) for 1 h in a shaker, the digestion solution was centrifuged twice at 1200 r/min for 12 min and cell suspension was filtered through 200 nylon meshes. The cells were resuspended in DMEM/F12 media with 10% FBS and penicillin/streptavidin for 10–14 days to allow attachment and the formation of colonies. These primary cultures were termed as passage 0. The medium was exchanged after 24 h to remove blood cells and medium exchange was performed every 2–3 days.
Adipogenesis of ASCs and Oil Red O staining
ASCs at the third passage were cultured in adipogenic inducer solution (DMEM/F12 containing 10% FBS, 1 µmol/L dexamethasone, 10 µmol/mL insulin, 0.5 mmol/L 3-isobutyl-1-methyl xanthine, and 200 µmol/L indomethacin; Sigma, St. Louis, MO, USA) for 2 weeks. Cells in control group were cultured in DMEM/F12 containing 10% FBS for 2 weeks. The medium was exchanged every 3 days. After induction for 14 days, the capacity of adipogenesis of ASCs was determined by staining with Oil Red O solution (Oil Red O solution:distilled water = 3:2). After washed with PBS, the cells were fixed in 10% formaldehyde at 37°C for 30 min. Then, the cells were maintained with Oil Red O solution for 30 min. Then, the cells were visualized and photographed in an inverted microscope (TMS).
Osteogenesis of ASCs and Alizarin Red Staining of calcified nodules
ASCs at the third passage were cultured with DMEM/F12 containing 10% FBS, 0.1 µmol/L dexamethasone, 50 µg/mL L−(+)−ascorbic acid, 10 mmol/L β-sodium glycerophosphate, and 10 mmol/L vitamin D (Sigma) for 3 weeks. Cells in control group were cultured in DMEM/F12 containing 10% FBS for 3 weeks. The medium was exchanged every 3 days. After 21 days of induction, the cells were stained by alizarin red to detect osteogenesis. After washed with PBS, the cells were fixed in 10% formaldehyde at 37°C for 30 min. Then, the cells were maintained with alizarin red solution for 30 min. Then, the cells were visualized and photographed in a microscope magnification (TMS).
Lentiviral infection of ASCs
After the cells reached 30%–40% confluence, the supernatant was removed. Then, the cells were cultured with lentiviral solutions of LV-EGFP and LV-CNAK-3Flag-EGFP. Enhanced infection solution (Polybrene solution and Eni.s. solution; Invitrogen) was then added to enhance infection efficiency. After 12 h of incubation at 37°C, the lentiviral solution was changed for additional 72 h. Then, the images of the cells expressing EGFP were captured under a fluorescence microscope (Eclipse Ti; Nikon).
In vivo efficacy
The experiment was approved by the Ethics Committee of Medical and Biological Research, Medical School of Xi’an Jiaotong University. MDA-MB-231 cells (106) were injected subcutaneously into the right flank of female BALB/c nude mice (Shanghai Silaike Laboratory Animal Co., Ltd, Shanghai, China) weighing about 20 g. The mice were randomly divided into two groups (n = 15 per group) after the volume of the tumor exceeded 300 mm3: a part of them were paratumorly injected with CNAK-ASCs suspension (experiment group) every 3 days for 2 months, and the others were injected with saline. The volume was evaluated every 3 days through the formula: length × width 2 × 0.5. After 2 months, all the mice were sacrificed. Then, the tumors and the relative tissues were extracted and weighted. For further experiments of immunohistochemistry (IHC) and hematoxylin and eosin (H&E Staining), the tumor, liver, lung, and spleen tissues were paraffin-embedded.
The tumor paraffin sections were incubated with 3% H2O2 deionized water for 10 min and washed with PBS three times for 5 min. After incubated with 10% goat serum for 30 min, mouse anti-monoclonal CD31 antibody (Abcam), rabbit anti-monoclonal CD133 antibody (Abcam), matrix metalloproteinase (MMP)-2 antibody (Cell Signaling Technology, Danvers, MA, USA), and MMP-14 antibody (Cell Signaling Technology) were added with them at 4°C overnight. Then, the relative biotin-labeled anti-mouse IgG was incubated with sections at 37°C for 15 min. The sections were incubated with horseradish peroxidase–avidin enzyme working solution for 15 min and 3,3′-diaminobenzidine (DAB) for 5 min. Then, hematoxylin was added to stain the nuclei. The tumor, liver, lung, and spleen paraffin sections were stained with H&E.
Statistical analysis
All statistical analyses were performed using SPSS 18.0 software (SPSS, Inc., Chicago, IL, USA). All values are presented as the mean ± SD. The experiments with two treatment groups were tested by Student’s t-test, and more than two treatment groups were tested by univariate analysis of variance (ANOVA); p < 0.05 was considered to indicate a statistically significant difference. All the graphs were made using the GraphPad Prism (Version 5.02).
Results
The cytotoxic activity of CNAK and CNRK on HUVECs
First, the cytotoxic activity of CNAK and CNRK on HUVECs was evaluated by MTT assays. The cells were cultured with various concentrations of CNAK or CNRK (0, 25, 50, 75, 125, and 200 µg/mL) for 24, 48, and 72 h. As shown in Figure 1(a), CNAK induced dose-dependent cytotoxicity in HUVECs. The proliferation of HUVECs treated with CNAK was significantly inhibited at the >50 µg/mL concentration (p < 0.05). However, the cytotoxicity decreased after 48 h. On the contrary, the significant cytotoxicity of CNRK was found at the >200 µg/mL (p < 0.05). Then, the integrity of HUVECs membrane was examined. As shown in Figure 1(b), CNAK induced cell membrane integrity loss, content outflow, and cellular necrosis.

The cytotoxic activity of CNAK and CNRK on HUVECs. (a) The cytotoxicity of CNAK and CNRK on HUVECs. The cells were cultured with various concentrations of CNAK or CNRK (0, 25, 50, 75, 125, and 200 µg/mL) for 24, 48, and 72 h. The cytotoxic activity was tested by MTT. CNAK induced dose-dependent cytotoxicity in the HUVECs. The proliferation of HUVECs treated with CNAK was significantly inhibited (p < 0.05; OD value: optical density). Data are presented as mean ± SD from triplicate determinations (*p < 0.05 versus control). (b) HUVECs were cultured with CNAK and CNRK for 48 h and observed under an inverted microscope (200× magnification). CNAK induced cell membrane integrity loss, content outflow, and cellular necrosis. (c) The effect of CNAK and CNRK on apoptosis of HUVECs. The cells were cultured with CNAK (100 µg/mL) and CNRK (100 µg/mL) for 24 and 48 h. The apoptosis of HUVECs was analyzed using flow cytometry for Annexin V/PI stain. The sum of the upper right and lower right quadrants is expressed as the percentage of the apoptotic cells. CNAK increased the proportion of apoptotic cells of HUVECs (p < 0.05). The results are represented as mean ± SD from triplicate determinations (*p < 0.05). (d) The expression of Caspase-3 and Bcl-2 was analyzed by western blot assays. The whole protein of HUVECs treated with CNAK (100 µg/mL) and CNRK (100 µg/mL) for 24 h were extracted. CNAK inhibited the expression of Bcl-2 and promoted Caspase-3 compared with CNRK and control. Data shown are representative images from three separate experiments.
The effect of CNAK on apoptosis of HUVECs
To determine whether apoptosis was the key point in the cytotoxicity of CNAK, Annexin V/PI double notation assay was applied. The cells were cultured with CNAK or CNRK at the same concentration (100 µg/mL) for 24 and 48 h. As shown in Figure 1(c), CNAK increased the proportion of apoptotic cells of HUVECs in a time-dependent manner (p < 0.05). In addition, the effect of CNAK on the expression of apoptosis-related proteins Caspase-3 and Bcl-2 was also tested by western blot (Figure 1(d)). It was found that the expression of Bcl-2 decreased and Caspase-3 increased in cells treated with CNAK compared with control (p < 0.05).
The expression of CD13 and the uptake of CNAK in different cell lines
The relative levels of CD13 expression in HUVECs, MCF-7, MDA-MB-231, L929, and HK-2 cells were first determined by western blot assays. As shown in Figure 2(a), the CD13 expression was detected in HUVECs and HK-2, while not in MCF-7, MDA-MB-231, or L929. Then, the uptake of CNAK in two CD13+ cell lines and a breast cancer cell line (MDA-MB-231) was assessed. The cells were incubated with CNAK (20 µg/mL) for 24 h, which is labeled by Rhodamine B in the K residue at C terminal of the amino acid. Then, the uptake of CNAK was assessed by a fluorescence microscope. As shown in Figure 2(b), the uptake of CNAK was evident in HUVECs, but not in HK-2 or MDA-MB-231 cells. Additionally, the fluorescent signals of HUVECs treated with CNAK (20 µg/mL) or CNRK (20 µg/mL) for 12 h were detected. It was found that both CNAK and CNRK could be took into by HUVECs (Figure 2(c)).
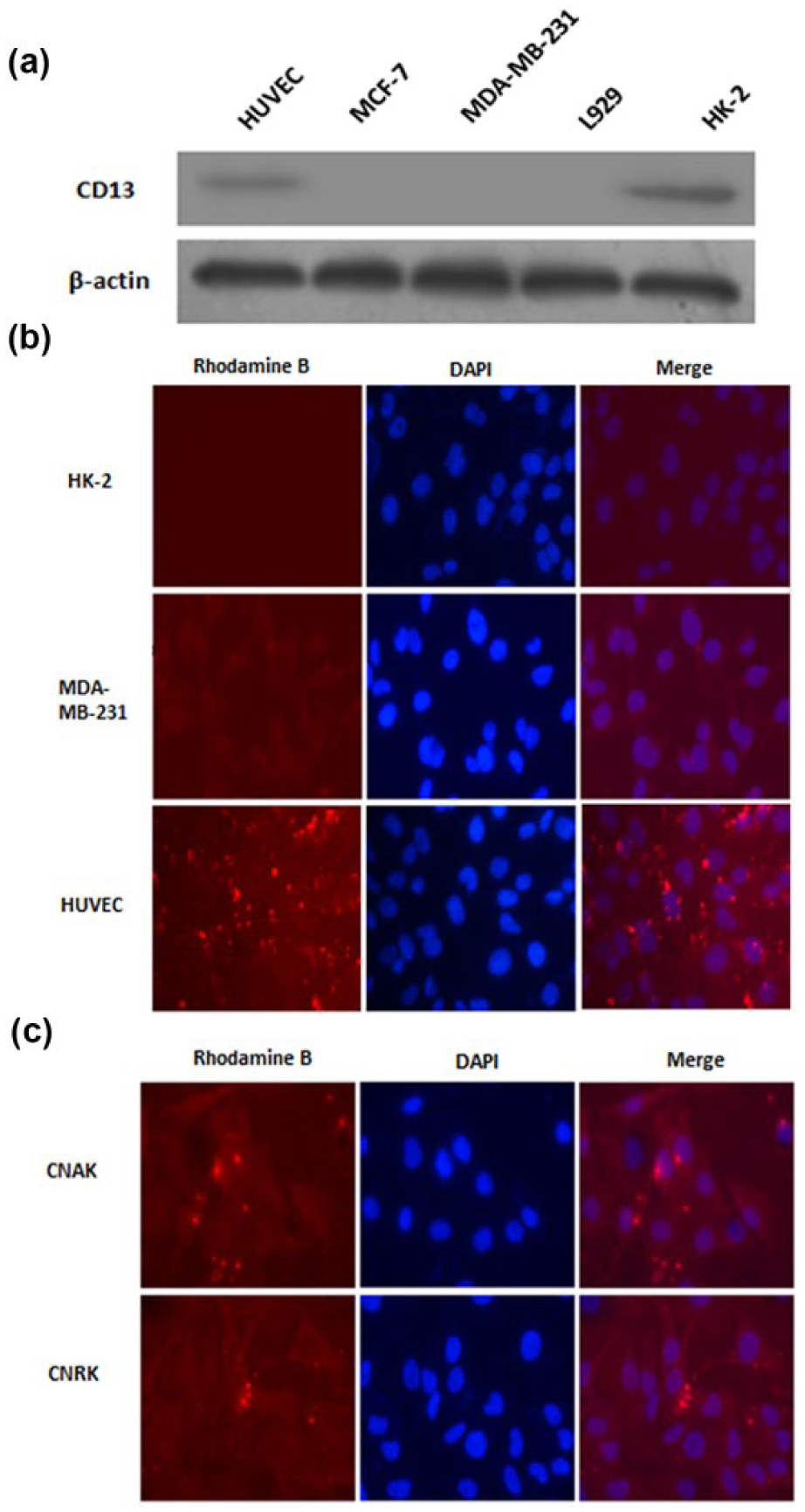
The expression of CD13 and the uptake of CNAK in different cell lines. (a) The expression of CD13 in different cell lines. The relative levels of CD13 expression in HUVECs, MCF-7, MDA-MB-231, L929, and HK-2 cells were determined by western blot assays. The CD13 expression was detected in HUVECs and HK-2, while not in MCF-7, MDA-MB-231, or L929. Data shown are representative images from three separate experiments. (b) Fluorescence photograph of HUVECs, MDA-MB-231, and HK-2 (200× magnification). The cells were incubated with CNAK (20 µg/mL) for 24 h, which is labeled by Rhodamine B in the K residue at C terminal of the amino acid. Then, the uptake of CNAK was assessed by a fluorescence microscope (200× magnification). The uptake of CNAK was evident in HUVECs, but not in HK-2 cells or MDA-MB-231 cells. Data shown are representative images from three separate experiments. (c) Fluorescence photograph of HUVECs treated with CNAK and CNRK for 12 h (200× magnification). The fluorescent signals of HUVECs treated with CNAK (20 µg/mL) or CNRK (20 µg/mL) for 12 h were detected. It was found that both CNAK and CNRK could be uptaken by HUVECs after incubated for 12 h. Data shown are representative images from three separate experiments.
The effect of CNAK on migration and tube formation of HUVECs
The effect of CNAK on migration and tube formation of HUVECs was analyzed by Transwell assays and wound healing assays. HUVECs were treated with normal media, CNAK (30 µg/mL), and CNRK (30 µg/mL) for 24 h in the upper chamber. The migrated cells were imaged and quantitatively analyzed. As shown in Figure 3(a), CNAK significantly inhibited the migration of HUVECs compared with the control group and CNRK (p < 0.05). The number of the migrated cells treated with CNAK (41.60 ± 4.19) was significantly less than that of the control (79.60 ± 3.27, p < 0.05) and CNRK (60.20 ± 3.15, p < 0.05). It was also found in wound healing assays that cells treated with CNAK hardly closed the scratch compared with the control and CNRK (p < 0.05, Figure 3(b)), which was consistent with the results of Transwell assays.

The effect of CNAK on cell migration and tube formation of HUVECs. (a) The effect of CNAK on cell migration of HUVECs evaluated by Transwell assays (200× magnification). HUVECs were treated with normal media, CNAK (30 µg/mL), and CNRK (30 µg/mL) for 24 h in the upper chamber. The migrated cells were imaged and quantitatively analyzed. The number of the migrated cells treated with CNAK was significantly less than that of the control and CNRK (p < 0.05). The data are mean ± SD from triplicate determinations (*p < 0.05). (b) The effect of CNAK on cell migration evaluated by wound scratch assays (40× magnification). HUVECs were cultured with normal media, CNAK (30 µg/mL), and CNRK (30 µg/mL) and the migration was determined by wound scratch assays. HUVECs treated with CNAK hardly closed the scratch compared with the control and CNRK (p < 0.05). The data are mean ± SD from triplicate determinations (*p < 0.05). (c) The effect of CNAK on tube formation of HUVECs (40× magnification). After incubation for 6 h with CNAK (25 and 50 µg/mL), the tubes formed by HUVECs were clearly observed under a microscope. The tube formation of HUVECs was significantly inhibited by CNAK compared with the control and CNRK (p < 0.05). The data are mean ± SD from triplicate determinations (*p < 0.05).
Then, the effect of CNAK on tube formation of HUVECs was assessed. As shown in Figure 3(c), after incubation for 6 h with CNAK (25 and 50 µg/mL), the tubes formed by HUVECs were clearly observed under a microscope. The tube formation of HUVECs was significantly inhibited by CNAK compared with the control and CNRK in a concentration-dependent manner (p < 0.05). The number of the branches from the tubules formed by HUVECs was counted by Image-Pro Plus software.
Isolation, culture, identification, differentiation, and the lentiviral infection of ASCs
Primary ASCs were isolated and cultured in vitro. As shown in Online Resource 1a, isolated cells in DMEM/F12 medium after 24-h cultivation under the light microscope were transparent, round, or oval in morphology, adhered to the plastic surface; 6 days after seeding, the number of the cells was increased and the cells with spindle-shaped morphology were observed. After 13 days of incubation, the spindle-shaped cells presented turbulence arrangement and cell fusion reached to 80%–90%. Following passage, the growth rate of the cells was increased. The growth curve of primary ASCs liked “S” shape by MTT, indicating strong in vitro expansion and proliferative potential.
Then, the isolated cells were identified by cell surface markers. Previous studies have reported that ASCs expressed the surface makers CD29, CD44, CD105, CD49b, CD49e, and CD90, but did not express CD31, CD34, CD45, HLA-DR, and CD133. 17 Additionally, Zuk et al. 3 revealed that ASCs expressed CD49d but not CD106, while BSCs expressed CD106 but not CD49d. It was found in this study that the cultured cells expressed CD29 and CD90, but did not express CD31, CD45, or CD106 (Online Resource 1b). It was indicated that we successfully isolated and cultured the ASCs.
Then, adipogenic induction and osteogenic induction of ASCs were assessed. As shown in Online Resource 1c and d, after cultured with adipogenic inducers and osteogenic inducers for 14 and 21 days, respectively, ASCs were stained with Oil Red O and Alizarin Red to label lipid droplets and calcium nodules, respectively. By contrast, no small lipid droplets or calcium deposition were observed in cells not treated with the adipogenic or osteogenic inducers.
As we mentioned above, ASCs are good carriers for transporting anti-angiogenesis drugs to cancer tissues because they can migrate to tumor tissues. Therefore, stably transfected ASCs which express CNAK peptide (CNAK-ASCs) were constructed and then validated by fluorescence microscope and western blot. As shown in Online Resource 1e and f, EGFP was highly expressed in ASCs transfected with the lentivirus. Besides, CNAK-3Flag-EGFP was detected at the expected size by western blot analysis in LV-CNGRC-3Flag-EGFP transfected cells. By contrast, no EGFP signal was detected in non-transfected cells and no expected peptide was detected in non-transfected or LV-EGFP-transfected cells. Therefore, stably transfected ASCs which express CNAK peptide were constructed.
The migration of CNAK-ASCs to breast cancer cells
To determine the potential of ASCs as carriers for transporting CNAK, the migration of ASCs to breast cancer cells was applied. ASCs were seeded in the upper chamber. Conditioned medium of MDA-MB-231, mouse fibroblast cell line L929, and normal DMEM/F12 medium containing 10% FBS were in the lower chamber. As shown in Online Resource 2a, ASCs were more powerful in migration toward breast cancer cells compared with other groups (p < 0.05). No obvious differences were found between fibroblast cells and normal DMEM/F12 medium. Next, the effect of lentiviral tranfection on the migration of ASCs was determined. There was no significant difference between before transfection and after transfection (Online Resource 2b). Therefore, ASCs can be considered as candidates for carrying CNAK.
Anti-tumor activity of CNAK-ASCs in vivo
Finally, the effect of CNAK-ASCs on the female athymic nude mice implanted with MDA-MB-231 cells was tested. The mice were randomly divided into two groups (n = 15 per group) after the volume of the tumor exceeded 300 mm3: a part of them was paratumorly injected with CNAK-ASCs suspension (experiment group) every 3 days for 2 months and the others were injected with saline (control group). The volume of tumors was evaluated every 3 days. The concentration of CNAK was referred from previous study. 18 As shown in Figure 4(a), the tumor volume of CNAK-ASCs group was significantly smaller than the control group (p < 0.05); 29 days after CNAK-ASCs injection, the tumor volume in CNAK-ASCs group (750 mm3) was reduced to 44% of the control group (1700 mm3). No abnormalities were observed in tumor, lung, liver, and spleen of mice in the histological examination (Figure 4(b)).
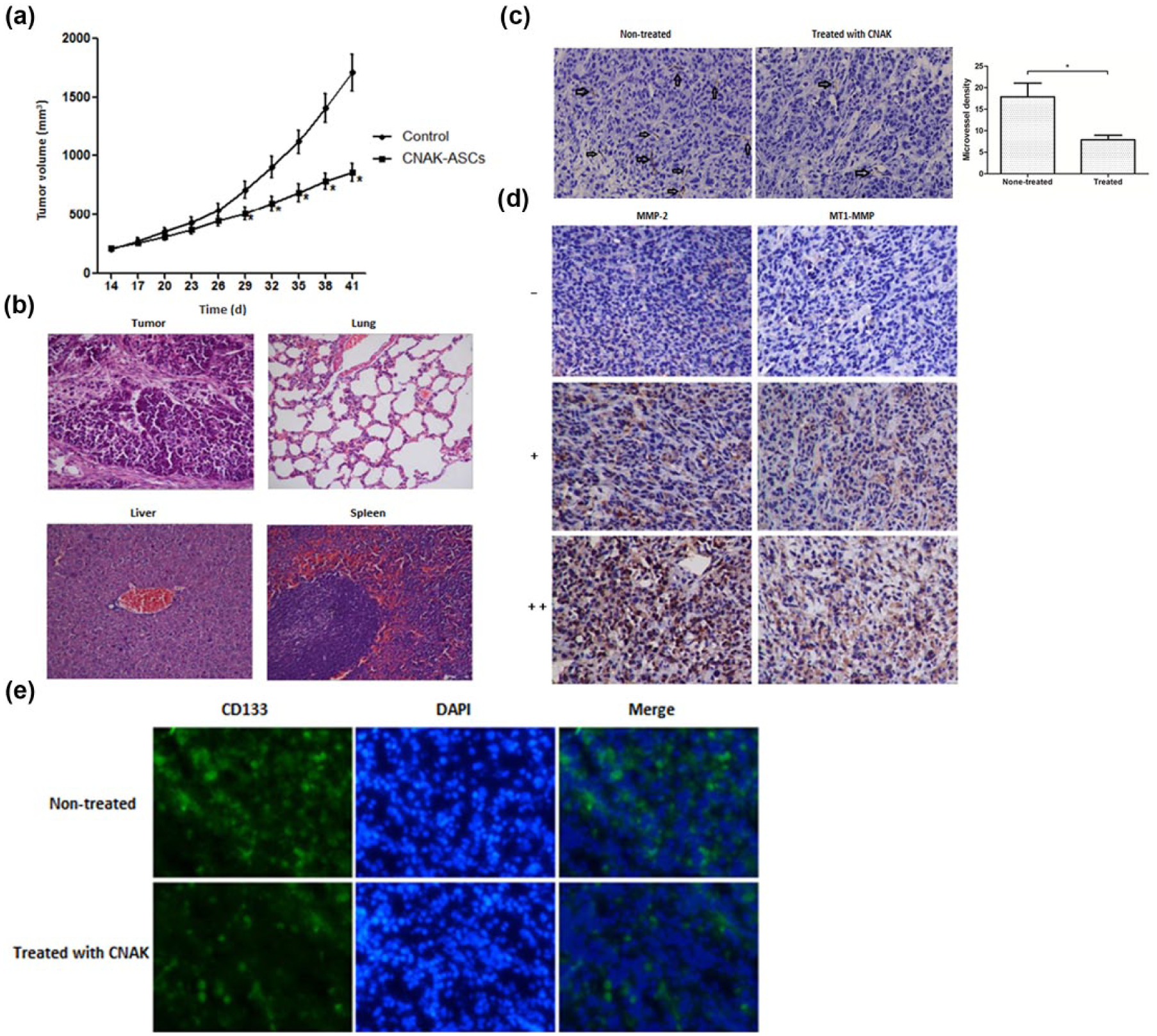
Anti-tumor activity of CNAK-ASCs in vivo. (a) The effect of CNAK-ASCs on tumor volume. Female athymic nude mice were implanted with MDA-MB-231 cells. The mice were randomly divided into two groups (n = 15 per group) after the volume of the tumor exceeded 300 mm3: a part of them were paratumorly injected with CNAK-ASCs suspension (experiment group) every 3 days for 2 months and the others were injected with saline (control group). The volume of tumors was evaluated every 3 days. The tumor volume of CNAK-ASCs group was significantly smaller than the control group (p < 0.05). (b) The images of the tumor, lung, liver, and spleen stained by hematoxylin and eosin (H&E) under the microscope (100× magnification). No significant damage was observed. (c) The effect of CNAK-ASCs on angiogenesis was assessed in tumor sections using immunohistochemistry (IHC; 400× magnification) with anti-CD31 antibody. The tumor microvessel density (MVD) was calculated using CD31 to mark vascular endothelial cells (arrows showed the positive staining). CNAK-ASCs significantly inhibited the angiogenesis in vivo (p < 0.05). (d) Representative photographs of the tumor sections examined by immunohistochemistry (IHC; 400× magnification). The expression of MMP-2 and MT1-MMP in two groups was examined through IHC analysis. The number of the stained cells was counted through five random fields in blinded manner. Every section was scored according to the staining intensity (the percentage of positively stained cells): weak express (+) represented the percentage of the positively stained cells ≤10%, strong positive expression (+++) indicated >60%, and moderate positive expression (++) indicated the percentage ranged from 10% to 60%. It was found that the expression of MMP-2 and MT1-MMP was significantly inhibited in CNAK-ASCs-treated group compared with the control group (Tables 1 and 2; p < 0.05). (e) The effect of CNAK-ASCs on the quantity of breast cancer stem cells (BCSCs; 100× magnification). The IHC assay for CD133 staining on tumor sections revealed that the number of CD133+ breast cancer cells in CNAK-ASCs-treated group was significantly less than saline-treated group (p < 0.05). Data are representative images and presented by mean ± SD of each group (n = 5 per group; *p < 0.05).
The effect of CNAK-ASCs on angiogenesis was assessed in tumor sections by IHC (400× magnification) with anti-CD31 antibody. The tumor microvessel density (MVD) was calculated using CD31 to mark vascular endothelial cells. As shown in Figure 4(c), the expression of CD31 was significantly inhibited by CNAK-ASCs compared with the control group (p < 0.05). Therefore, CNAK-ASCs inhibited the angiogenesis in vivo.
MMP-2 and MT1-MMP secreted by endothelial cells and cancer cells were important in decomposing matrix and vessels. The expression of MMP-2 and MT1-MMP in breast cancer tissues was related to angiogenesis. Therefore, the expression of MMP-2 and MT1-MMP in two groups was examined by IHC analysis. Representative photographs were seen in Figure 4(d). The number of the stained cells was counted through five random fields in blinded manner. Every section was scored according to the staining intensity (the percentage of positively stained cells): weak express (+) represented the percentage of the positively stained cells ≤10%, strong positive expression (+++) indicated >60%, and moderate positive expression (++) indicated the percentage ranged from 10% to 60% (Figure 4(d)). As shown in Tables 1 and 2, the positive rate of the expression of MMP-2 and MT1-MMP in CNAK-ASCs-treated group was 33.3% (5/15) and 26.7% (4/15), respectively. In control group, the positive rate of the expression of MMP-2 and MT1-MMP was 66.7% (10/15) and 60% (9/15), respectively. The expression of MMP-2 and MT1-MMP in CNAK-ASCs group was significantly inhibited compared with the control group (p < 0.05).
The number of positive cases for expression of MMP-2 in the tested samples of two groups.

MMP-2: matrix metalloproteinase-2.
The number of positive cases for expression of MT1-MMP in the tested samples of two groups.

MT1-MMP: membrane type 1-matrix metalloproteinase.
Then, the effect of CNAK-ASCs on breast cancer stem cells (BCSCs) in breast cancer xenografts was studied. The IHC assay for CD133 staining on tumor sections revealed that the number of CD133+ breast cancer cells in CNAK-ASCs-treated group was significantly less than saline-treated group (p < 0.05), indicating breast cancer stem cells were significantly inhibited by CNAK-ASCs (Figure 4(e)).
Discussion
CD13 has been reported overexpressed in newly formed tumor vessels and some tumor cells. 19 It is reported that various bioactive molecules can be combined or integrated with the NGR motif to recognize CD13 and promote antitumor activity. 20 Therefore, targeted drugs containing NGR motif are important in the treatment of tumors. In this study, a new peptide (CNAK) containing the cyclic NGR motif was designed and its biological effect on CD13+ HUVECs was studied. It was found that CNAK had an anti-angiogenic effect on breast cancer in both in vitro and in vivo experiments. The synthesized peptides CNAK containing pro-apoptotic structure presented dose-dependent cytotoxicity in HUVECs. The peptide induced apoptosis of HUVECs via enhancing the apoptosis-related protein Caspase-3 and inhibiting the expression of Bcl-2. It is reported that the cytotoxicity of cyclic NGR motif combined with pro-apoptosis peptides tumor necrosis factor (TNF) is stronger than cyclic NGR or TNF alone, which is similar to our study. 21
Drugs containing NGR motif mainly interact with cells that positively express CD13 and enter cells depended on endocytosis. In this study, the levels of CD13 expression in HUVECs, MCF-7, MDA-MB-231, L929, and HK-2 cells were determined by western blot. Then, the uptake of CNAK in different cell lines was detected. Both CNAK and CNRK could be uptaken by CD13+ HUVECs but not by CD13+ HK-2 cells. However, in CD13-MDA-MB-231 cells, a few of fluorescent signals were also observed. It might be related with the integrins on the surface of the cells. It is reported that αυβ3 and αυβ5 which can bind to NGR motif are mainly overexpressed on these cells. 22 Cancer angiogenesis is involved with some biological behaviors of vascular endothelial cells, such as migration and differentiation. In this study, the migration and tube formation of HUVECs were significantly inhibited by CNAK compared with the control and CNRK. The stronger cytotoxicity and inhibition of CNAK against HUVECs than CNRK may derive from particular structure with pro-apoptotic function of CNAK.
Current studies have revealed that ASCs and BSCs have similar phenotypes and differentiation potentials. However, ASCs are easily obtained, cultured, and transfected in vitro and can survive following transplantation into animals. 23 Compared with bone marrow tissue, adipose tissue has tremendous advantages: first, for human and other mammals, adipose tissue is ubiquitous and easily isolated. Second, adipose tissue contains large amounts of mesenchymal stem cells. Under a local anesthesia, only 1.2 × 105 stem cells from 40 mL of bone marrow can be obtained. However, 4 × 106 stem cells can be obtained from 200 mL of adipose tissue under the same condition. The yield of the stem cells can be increased by almost 30 times. In order to get a large amount of bone mesenchymal stem cells, the patients must undergo surgical operation and suffer general anesthesia. Third, isolation of adipose tissue is easier than that of bone marrow with minimal invasive procedure and complications. 24 Finally, it is reported that ASCs are more efficiently infected by lentiviral vectors than BSCs.25,26 Therefore, ASCs are proper candidates which can be used as antitumor agents in application of stem-cell-based therapies and tissue engineering. In this study, ASCs from rat adipose tissue were successfully isolated, and stably transfected ASCs which express CNAK peptide (CNAK-ASCs) were constructed. ASCs were used to localize CNAK on vascular endothelial cells in tumor tissues based on their homing property. It was found that ASCs owned an obvious migration toward breast cancer cells, and the transfection of CNAK gene had no significant influence on the migration of ASCs.
Based on the important role of angiogenesis for tumor progress, CD31 was used to stain tumor angiogenesis and the tumor MVD was assessed. As the results showed, a significant decrease in the expression of CD31 was observed in tumors treated with CNAK-ASCs compared with the control, that is, CNAK-ASCs significantly inhibited the tumor angiogenesis in vivo.
MMPs are zinc-dependent endopeptidases, recognized as poor prognosis markers for breast cancer patients. They can degrade extracellular matrix (ECM) and basement membrane components to integrate basement membrane.27,28 Recent studies have shown that MMPs are involved in tumor-induced angiogenesis. In particular, MMP-2, MMP-9, and MMP-14 (MT1-MMP) are crucial for tumor angiogenesis and the formation of metastasis. Therefore, they are considered as crucial targets in cancer therapy. 29 It is reported that 2-deoxyglucose (2-DG) inhibits angiogenesis with an action which is involved in the inhibition of vascular endothelial growth factor receptor 2 (VEGFR2) signaling and MMP-2 expression. 30 Additionally, MT1-MMP seems more important. It has been proposed that MT1-MMP plays a central role in tumor growth, invasion, and neovascularization. Besides cleaving matrix proteins, it can activate proMMP-2 which results in an amplification of pericellular proteolytic activity. Selectively blocking proMMP-2 processing on tumor and endothelial cells using DX-2400 (a highly selective fully human MMP-14 inhibitory antibody) is reported to inhibit angiogenesis and slow tumor progression and formation of metastasis. 31 Based on the above, the expression of MMP-2 and MT1-MMP in two groups was evaluated by IHC analysis. It was found that the expression of MMP-2 and MT1-MMP in CNAK-ASCs group was significantly inhibited compared with the control group (p < 0.05). The results indicated that angiogenesis was inhibited by CNAK-ASCs in vivo. Therefore, CNAK was transported to breast cancer tissues via the migration of ASCs, and its anti-angiogenesis effect was detected in vivo.
Currently, the failures to cure breast cancer are mainly because of the recurrence and metastasis. It is indicated that the existence of breast cancer stem cells (BCSCs) may result in the resistance to traditional therapies. 32 It is widely believed that BCSCs are only in a minority in tumor cells. Nonetheless, in triple-negative breast tumor cells (TNBC), the percentage of CD44+/CD24−/low cells is up to 90%. However, recent studies have reported that a portion of TNBC which highly expressed CD133 is more aggressive and it can enhance the expression of resistance-related proteins. 33 Therefore, CD133 may be a new marker for BCSCs of TNBC. In this study, the number of CD133+ breast cancer cells in CNAK-ASCs-treated group was significant less than saline-treated group (P < 0.05), indicating breast cancer stem cells were significantly inhibited by CNAK-ASCs.
In summary, a new peptide (CNAK) containing the cyclic NGR motif was designed and its biological activity against HUVECs was studied. Stably transfected ASCs which express CNAK peptide (CNAK-ASCs) were established and their anti-angiogenic property in vivo was investigated. ASCs were employed to localize CNAK on vascular endothelial cells in tumors based on their homing property. Our experiments provided that CNAK-ASCs may be valuable for targeting endothelial cells of tumors. Further studies to examine other specialized cell lines, the mechanisms of the anti-angiogenic activity of CNAK and its effect on breast cancer stem cells as well as their microenvironment should be performed in the future.
Footnotes
Acknowledgements
G.W. and N.Y. contributed equally to this work.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was funded by a grant from the National Natural Science Foundation of China (grant no. 81201680).