
Abstract
Current vascular targeting strategies pursue two main goals: anti-angiogenesis agents aim to halt sprouting and the formation of new blood vessels, while vascular disrupting agents along with coaguligands seek to compromise blood circulation in the vessels. The ultimate goal of such therapies is to deprive tumor cells out of oxygen and nutrients long enough to succumb cancer cells to death. Most of vascular targeting agents presented promising therapeutic potential, but the final goal which is cure is rarely achieved. Nevertheless, in both preclinical and clinical settings, tumors tend to grow back, featuring a highly invasive, metastatic, and extremely resistant form. This review highlights the critical significance of tumor rim cells as the main factor, determining therapy success with vascular targeting agents. We present an overview of different single and combination treatments with vascular targeting agents that enable efficient targeting of tumor rim cells and long-lasting tumor cure. Understanding the nature of tumor rim cells, how they establish, how they manage to survive of vascular targeting agents, and how they contribute in tumor refractoriness, may open new avenues to the development of beneficial strategies, capable to eliminate residual rim cells, and enable tumor ablation once and forever.
Keywords
Introduction
Mode of action of vascular targeting agents
In recent years, targeting the tumor cell–specific markers is widely applied in oncology practice.1,2 Although these therapies resulted in beneficial effects for subsets of tumors, the ultimate goal, which is cure, is very rarely accomplished. 3 However, the unique characteristics of tumor vasculature confer it an ideal target for the development of selective vascular targeting agents (VTAs).4–7 Current VTAs split into three main groups, and through a different mode of action, they all aim to cut off tumor nutrient and oxygen path, to starve and suffocate tumor cells to death. 8
Inhibition of tumor neovascularization by anti-angiogenesis agents
The history of anti-angiogenesis approach goes back to 1971, when Judah Folkman reported the isolation of a pro-angiogenic tumor factor, and the same year recommended the exploitation of anti-angiogenic agents as anti-cancer drugs. 9 The purpose of anti-angiogenesis therapies is to halt generation of new blood vessels (sprouting)10–14 or alternatively to inhibit proliferation of tumor endothelial cells (ECs).15–18 Examples of different classes of these anti-angiogenesis agents (AAAs) are listed in Table 1. While most AAAs are reported to exacerbate hypoxia in tumor cells, anti–vascular endothelial growth factor (VEGF) antibodies, for example, bevacizumab, are reported to improve blood flow by decompression of blood vessels and normalizing tumor vasculature.
Different classes of anti-angiogenesis agents (AAAs).

HIF: hypoxia-inducible factor; mTOR: mechanistic target of rapamycin; VEGF: vascular endothelial growth factor; VEGFR: vascular endothelial growth factor receptor; aFGF: acidic fibroblast growth factor; bFGF: basic fibroblast growth factor; FGFR: fibroblast growth factor receptor; PDGFR: platelet-derived growth factor receptor; CSF-1R: colony stimulating factor 1 receptor; KDR: kinase insert domain receptor; EC: endothelial cell; EDA: extracellular domain A; EDB: extracellular domain B; MMP: matrix metalloproteinase.
RNA aptamer for VEGF-A.
Disruption of tumor blood vessels by vascular disrupting agents
In the early 1980s, Juliana Denekamp verified that tumor regression occurs when the blood flow is compromised. 19 Vascular disrupting agents (VDAs) exploit the distinct shape of tumor ECs to induce selective vascular shut down, without requiring a targeting moiety. There are two types of VDAs: flavonoids, including 5,6-dimethylxanthenone 4-acetic acid (DMXAA) and its synthetic derivatives, and tubulin-binding agents, for example, combretastatin A-4 phosphate, ZD6126, and Oxi4503. 20 The mode of action of flavonoids is to cause semi-disintergration of the actin cytoskeleton, division of DNA strands, and induction of EC apoptosis. 21 However, tubulin-binding agents disturb the ECs’ dependence on the tubulin cytoskeleton to retain their shape. 22
Infarction of tumor blood vessels by coaguligands
Myocardial/brain infarctions are the top leading causes of death around the globe. A single blood clot would be enough for congestion of a single capillary and subsequent death of a thousand cells. The same strategy could be adopted to combat cancer by generation of tumor-specific coaguligands. 23 Coaguligands consist of a truncated form of human tissue factor (tTF), which is fused to a tumor EC moiety. Upon binding to the tumor vasculature, TF induces selective thrombosis, leading to subsequent ischemia, and infarction of tumor blood vessels.24,25 In contrast to VDA therapy, TF-induced tumor vascular infarction has met with beneficial effects in eradication of large tumors in animal models, yet this approach suffers from incomplete induction of thrombosis, which leaves a ring of viable tumor cells at the tumor edge, similar to the effect of VDAs on large bulky tumors.26,27
Residual rim cells: the main barrier to successful VTA therapy
The unequal oxygen distribution found within solid tumors results in the establishment of three distinct layers of cells: (1) the anoxic center core composed of necrotic cells, (2) the hypoxic middle layer harboring dormant cancer cells, and (3) the normoxic outer layer featured by highly proliferative tumor cells (Figure 1). 28 The outer proliferative layer is a target for tumor-reducing agents, including chemotherapy and anti-angiogenesis, which elicit cytostatic effects on the highly proliferating cells at the tumor periphery. 29 However, the necrotic inner layer provides a heaven for localization of anaerobic bacteria. 30 The interstitial locations of cells in the middle layer, however, render them out of reach of most anti-cancer agents. More importantly, constant low-grade hypoxia in the middle layer selects for hypoxia-tolerant cells, known as tumor rim cells, extremely resistant cells to eradicate. 28 The most effective VDAs producing more than 90% widespread central necrosis still leave a thin ring of viable cells from which tumor revives. 20 It appears that the existence of tumor rim cells remains the foremost impediment to successful tumor ablation encountered by vascular targeting strategies, especially VDAs. 31 Although there is no evidence for the existence of tumor rim cells following anti-angiogenic treatment, tumor survival and aggressive phenotype encountering by AAAs justify the likelihood of the existence of such resistant population of tumor cells. The running hypothesis for the ineffectiveness of VDAs to eradicate tumor rim cells is that rim cells might nourish from surrounding normal vasculature; hence, they are immune to vascular disruption and hypoxia. 3 Although, potent VDAs are shown to induce widespread hypoxia, encompassing viable tumor edge cells, as well. 20 Furthermore, it is established that widespread thrombosis induced by TF fusion proteins leads to tumor vascular infarction, ischemia, and degeneration of tumor cells, as well as the tumor rim cells and cure.23,32 These observations suggest that hypoxia-tolerant tumor rim cells are not ordinary tumor cells to target, yet sustained deprivation of tumors out of oxygen and nutrients would suffice to succumb these tough cells to death. 32 As most VTAs are shown to induce a high grade of hypoxia, rather than anoxia, tumors are forced to divert to compensatory mechanisms, including switching to alternative angiogenic pathways,33,34 stimulating dormant cells, 35 selecting aggressive and metastatic cells,36,37 autophagy, 38 establishment of cancer stem cells,39,40 and recruiting vascular progenitor cells41,42 as well as tumor-associated macrophage (TAM).43–45 Features that tumor exploits to escape oddly shape the typical characteristics of challenging tumor rim cells as well. 28
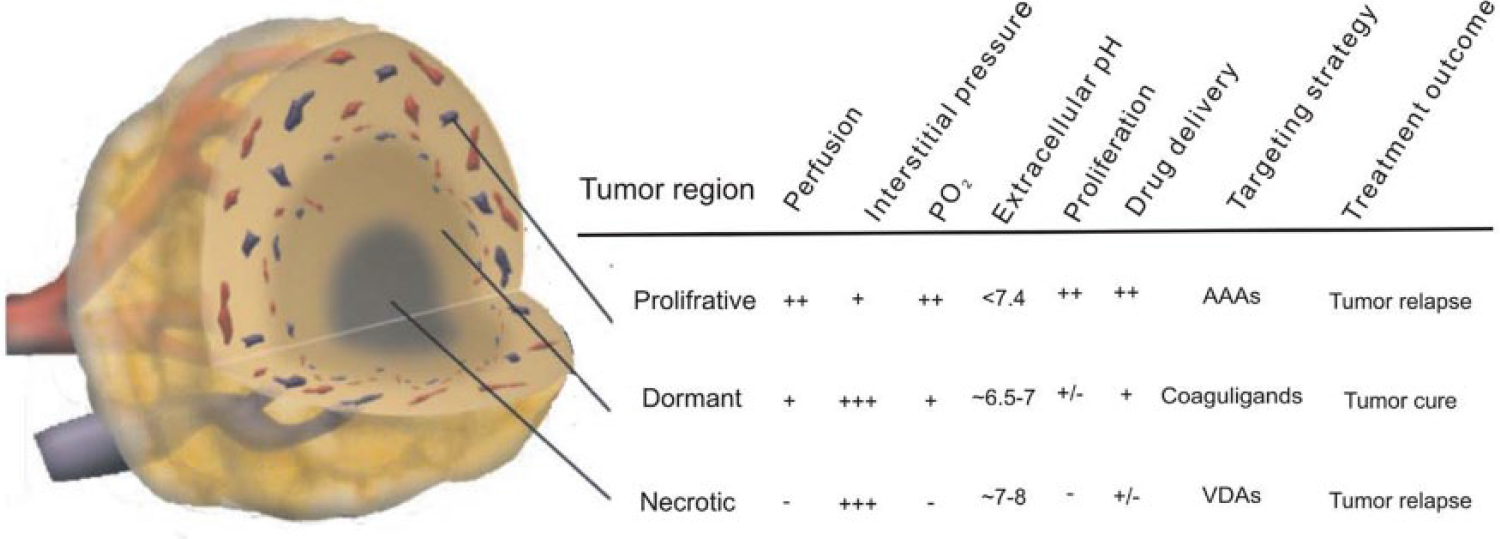
The characteristics of different layers of tumors. Spontaneous solid tumors are composed of three distinct layers of cells. The outer proliferative layer which is in close contact to the bloodstream and could be readily targeted by most anti-cancer drugs. The necrotic inner layer (tumor core) provides a heaven for propagation of anaerobic bacteria that feed on tumor cells/or alternatively deliver cytotoxic drugs to the tumor core. The interstitial location of cells in the middle layer, however, renders tumor cells out of reach of cytotoxic drugs that could attack tumor cells either from inside (tumor core) or outside (the tumor periphery). Furthermore, adaptation of tumor cells to the lower O2 tension in the middle layer selects for the dormant aggressive cells–the rim cells—hard to target and eradicate by current anti-cancer approaches. Rim cells occasionally could be targeted by coaguligands. Smart combination rational, however, could increase the chance of targeting rim cells and enable tumor cure.
Eradication of tumor rim cells and complete remission of tumor by single/combination therapies with VTAs
Eradication of tumor rim cells by VTAs (single modalities)
Among different vascular targeting strategies, several approaches reported to elicit curative potentials as single therapies in preclinical settings. Complete tumor ablation by overcoming tumor rim cells is accomplished with ligand-directed vascular targeting, coaguligands, DNA vaccines, and photodynamic therapy (PDT; Table 2).
Complete tumor ablation by single/combination therapy with VTAs.

TV: tumor volume; i.p.: intraperitoneal; i.v.: intravenous; s.c.: subcutaneous; DMXAA: 5,6-dimethylxanthenone 4-acetic acid; MMC: mitomycin-C; cDNA: complementary DNA; FAA: flavone-8-acetic acid; A5B7: anti-carcinoembryonic antigen IgG; CR: complete remission; q: interval between injections; “×”: number of repeated doses; PSMA: prostate-specific membrane antigen; tTF: truncated form of human tissue factor.
Ligand-directed vascular targeting
As a potent anti-angiogenic strategy, ligand-directed vascular targeting is recognized as a worthy anti-cancer approach. For this, ligand could be a monoclonal antibody (mAb)/molecule against a proliferation-associated antigen on ECs and the VTA could be a toxin (immunotoxin (IT)),50,51,82–84 cytokine (immunocytokine (IC)),52–61,63,85–89 apoptotic, cytotoxic (antibody–drug conjugate (ADC)),54,90,91 photosensitizer (PS),92,93 radioisotopes (radionuclides and radiotracers),94–96 or coagulation factor (coaguligand).23,32,77–81,97 Examples of different ligand-directed vascular targeting are listed in Table 3. When targeted to tumor vasculature, ligand-directed VTA inhibits tumor-associated angiogenesis and destroys tumor vasculature by selectively reacting with proliferating ECs. 54 However, activated/proliferating ECs facilitates endocytosis of the fusion protein inside tumor cells, where the internalized payloads could lead to further cytotoxicity. Matsuno et al. 51 developed anti-endoglin mAbs conjugated to the deglycosylated ricin A (dgRA) chain and tested their anti-tumor efficacy for treating MCF-7 breast carcinoma xenografts in mice. Long-lasting complete regression of the tumors was induced, when three doses of 40 µg of the ITs were administered. In a similar study by Seon et al., 50 long-lasting complete remission (CR) was accomplished in all animals (100%), which were given three doses of mAb-dgRA.
Different ligand-directed VTAs.

MMAE: monomethyl auristatin E; tTF: truncated form of human tissue factor.
SAA: 7-amino-
Tumor vascular infarction
Induction of selective thrombosis in tumor vasculature by TF fusion proteins (coaguligands) leads to severe ischemia, and in case of the maintained complete thrombosis, it could lead to the eradication of tumor rim cells and cure. Huang et al. 32 generated the first coaguligand to induce selective tumor vascular infarction in neuroblastoma tumors in mice. A bispecific Ab composed of tTF and Ab against MHC-II—an artificial marker of angiogenesis—was injected into mice bearing large C1300 (Muy) tumors. Upon a single injection, massive thrombosis occurred throughout tumor in 30 min, and extensive intravascular thrombosis of the tumor lasted for more than 72 h, resulting in complete regressions in 38% of the treated mice, lasting 4 months or more. Partial tumor regression in mice was consistent with undetectable MHCII antigens in the tumor periphery and incomplete induction of thrombosis by the coaguligand, which allowed rim cell survival. Nilsson et al. 79 targeted tTF to EDB domain of fibronectin on vasculature of three different types of solid tumors. Although a single 20 µg injection of the ScFv (L19)-tTF induced the rapid infarction of large tumors, this dose of coaguligand failed to eradicate tumor rim cells, and tumor tends to grow from a residual ring of viable cells close to the peritoneum. Higher doses of coaguligand (35 µg) enabled complete ablation of residual tumor mass and lead to CR in ~30% of the treated mice. Eradication of tumors by other coaguligands, including Ttf–prostate-specific membrane antigen (PSMA), tTF-VEGF, and tTF-NGR, is reported by other groups as well.77,80,81
DNA vaccines
Given the fact that tumors can continuously undergo mutations to alter their antigenic features, they can evade from host immune responses and become resistant to the anti-cancer therapies.76,98 Tumors are immunosuppressive in a variety of ways, and pro-angiogenic cytokines and their receptors, including VEGF family, fibroblast growth factor (FGF), and matrix metalloproteinases (MMPs), elicit central role in these immunosuppressive events.99,100 For example, VEGF may functionally inhibit the immune system, partly by preventing the maturation of dendritic cells and inhibiting early T-cell development. 101 In this regard, immune-based cancer therapies that target angiogenesis may potentiate a broader anti-tumor immune responses in various ways: (a) hamper angiogenesis via blocking pro-angiogenic cytokines; (b) ameliorate the tumor immunity by interfering with immunosuppressants; (c) target multiple angiogenic mediators, which enhances the effectiveness of anti-angiogenesis; (d) circumvent the need for repeated drug administration; and (e) moreover, immunotherapy could exert not only therapeutic but also prophylactic potential by suppression/eradication of metastasis.102–105 In the study by Teramoto et al., 62 pigeon cytochrome c–derived peptides (Pan-IA) containing dendritic cells were immunized in combination with ovalbumin (OVA) antigen DNA to prime both T helper and cytotoxic T lymphocyte (CTL) immunity in mice bearing tumor xenografts. Pan-IA is shown to trigger T helper function in mice, through efficient binding to a wide variety of MHC class II molecules. In this study, tumor-specific cytotoxicity and helper immunity were only primed in the combination setting and resulted in augmented tumor lysis ability, interferon (IFN)-gamma-mediated anti-angiogenic effects, and CR of tumor in ~70% of the treated mice.
PDT
PDT is now largely appreciated in treatment of tumors, especially the superficial ones. This modality incorporates a visible light to convert nontoxic oxygen molecules within tumors into cytotoxic singlet oxygen, through a PS. The antibody–PS conjugate could be employed for selective directing of PS to the tumor site. 93 PDT consumes tumor oxygen supply to generate reactive oxygen molecules (ROS) and to induce apoptosis/necrosis of tumor cells. In addition, PDT could be formulated to destroy tumor vasculature as well. 106 Palumbo et al. 67 verified the efficacy of PS-L19 for selective targeting of tumor neovasculature in mice bearing F9 murine teratocarcinoma and A431 human epidermoid carcinoma xenografts. L19 motif allowed specific localization of PS on tumor vasculature in both tumor models in mice. Upon irradiation, porphyrin-based PS promoted PDT and permitted selective tumor vascular disruption, leading to complete and long-term tumor eradication. Furthermore, depletion studies indicated that natural killer (NK) cells were indispensable for lifelong complete responses.
As single modality, a perfect VTA should be able to penetrate deep into the hypoxic regions, where dormant rim cells reside. In addition, such therapy should provide a sustained toxic dose of VTAs enough to kill any residual cancer cells. While most VTAs render a narrow therapeutic dose and fail to provide a higher retention time, combination therapies are indicated to overcome VTA-associated resistance in tumors.
Eradication of tumor rim cells by VTAs (combination therapies)
Most of current VTAs in the clinic need to couple with other anti-cancer modalities; the concerted work of two different agents leads to the enhanced therapeutic potential and enable tumor remission. The aim of such combination therapy is to improve the odds of successful targeting of the rim cells, which lower the risk of subsequent tumor survival and invasion and consequently enable tumor eradication for good. Notably, drug timing and sequencing is of critical significance determining the curative potential for combination therapies with VTAs.
AAAs combined with AAAs/chemotherapy
Given to the redundancy in angiogenic factors, the concept for concurrent application of two different AAAs is clear. Nonetheless, the rational for combination of AAAs with chemotherapy is to improve delivery of chemotherapeutic agents, as the first class of AAAs is shown to improve tumor blood flow by decompression of tumor blood vessels; hence, administration of AAAs prior to intravenously (i.v.) administered chemotherapeutic agents would lead to improved uptake of chemotherapeutic agents by tumor cells. Cao et al. 46 evaluated the anti-tumor activity of bevacizumab combined with topoisomerase I inhibitor irinotecan (CPT-11) against human head and neck squamous cell carcinoma (HNSCC) xenografts in mice. Daily bevacizumab therapy was initiated 1 week in advance to CPT-11 treatment and continued for 4 weeks. Monotherapy with irinotecan resulted in almost 40% CR, while bevacizumab alone showed no curative potential. Combination therapy of bevacizumab with irinotecan produced marked inhibition of FaDu tumor growth and resulted in 80% CR of tumor in mice. Drug scheduling was found crucial for effective chemotherapeutic effects since bevacizumab potentiated irinotecan curative potential, only when lower doses of bevacizumab administered 7 days prior to irinotecan. Concurrent administration of both drugs and/or injection of higher doses of bevacizumab failed to potentiate anti-tumor activity of irinotecan. In another study, Dings et al. 47 reported combination of anginex (angiostatic agent) and 0118 (topomimetic of anginex) with chemotherapeutic irofulven to treat human ovarian tumor xenografts in mice. The combination of a low dose of irofulven with either AAAs was more effective at inhibiting tumor growth than any of the monotherapies, as the anginex/irofulven and 0118/irofulven combinations produced 92% and 96% tumor growth inhibition, respectively. The 0118/irofulven combination resulted in 100% complete responses in treated mice. A combination of AAAs with low-dose irofulven not only augmented the anti-tumor efficacy of irofulven but also abolished administration of toxic doses of this chemotherapeutic in single therapy. In an additional study, Volk et al. 48 explored the anti-tumor and anti-metastatic potential of bevacizumab combined with nab-paclitaxel (an albumin-bound form of paclitaxel) in mice bearing orthotopic breast tumors. Mice bearing luciferase-tagged MDA-MB-231 tumors were treated with one, two, or three cycles of bevacizumab, nab-paclitaxel, or combination of both. Bevacizumab therapy abrogated the VEGF-A protective effect upon nab-paclitaxel cytotoxicity and restored breast tumor sensitivity to chemotherapy. Combined work AAA and chemotherapeutic nab-paclitaxel markedly inhibited tumor growth and metastasis, resulting in up to 40% CR. This therapy was also promising in reduction of both lymphatic and pulmonary metastases by 60% and 100%, respectively. Burrows et al. 49 examined the single and combined therapeutic potential of EC IT (anti MHCII) M5/114-dgA and anti-tumor IT 11-4.1-dgA (anti MHCI) in large neuroblastoma tumors. In a dose-dependent fashion, low doses of M5/114-dgA (20 µg) gave negligible and transitory responses in animals, whereas high doses (40 µg) of IT resulted in marked regression of tumors in all animals, but the responses were temporary, since tumors started to repopulate 1 week after. A combination of two ITs led to long-term eradication of tumors in 5/8 of the animals. In view of the fact that the efficacy of rituximab-based therapies is largely relied upon the extent of ADCC-mediated destroying of lymphoma cells, Schliemann et al. adopted different single and combination treatment schedules of rituximab (mAb against CD20 on the surface of B cells) and L19-IL2 in CB17/lcr SCID mice bearing Ramos and DoHH-2 human B-cell lymphoma xenografts. In Ramos tumor xenografts, CR of tumor (4/5 mice) was observed, when 200 µg rituximab combined with 6.6 or 20 µg L19-IL2; however, long-lasting CR for more than 3 months was observed with higher doses of L19-IL2 (4/5 mice). The results were reproducible, and in DoHH-2 follicular lymphoma xenografts, 200 µg rituximab + 20 µg L19-IL2 led to additive effects and long-lasting CR in all animals (5/5 mice). Gutbrodt et al. 54 treated immunocompetent mice bearing acute myelogenous leukemia (AML) tumors with a combination of cemadotin thiol or IL2 as cytotoxic drugs containing F8 as a vascular targeting motif. Anti-hen egg lysozyme Ab KSF was served as a negative control. When tumors reached to the volume of 50–100 mm3, mice were given i.v. injections of either saline, F8-IL2 (1 mg/kg), F8-SSCH2Cem (10 mg/kg), KSF-SS-CH2Cem (10 mg/kg), or F8-IL2 + F8-SS-CH2Cem. Combination of F8-SS-CH2Cem and F8-IL2 resulted in long-lasting complete tumor eradication in 4/5 mice. Furthermore, upon rechallengement with C1498 cells after ~4 months, all cured animals rejected tumors, indicating the acquired protective immunity in this setting. Schwager et al. 53 evaluated the efficacy of L19–IL2 in combination with a murine anti-CTLA-4 antibody (a mouse analog of Ipilimumab) or with L19-TNF in F9 murine teratocarcinomas and CT26 colon carcinoma xenografts. CR was demonstrated in both tumor models, when L19–IL2 was combined with CTLA-4 blockade. Since in F9 tumors the CTLA-4 blockage is not active and NK cells are the main regulators of therapeutic activity of targeted IL2, cured mice from F9 tumors developed lesions upon rechallenging with F9 tumor cells. In the CT26 model, T-cells are the main mediators accounting for anti-tumor action of IL2 and cured mice remained tumor free upon rechallenging with the parental cancer cells. Similarly, a single intratumoral injection of L19–IL2 induced CR once it coupled with L19-TNF therapy, whereas the two drugs failed to produce any cures as single modalities. Pasche et al. 59 explored therapeutic performance of IL-12 combined with IL2 and paclitaxel. In this study, the combination of F8-IL2 and mIL12-F8-F8 led to anti-angiogenic effects induced by activation of INFγ and CR in 2 of 3 mice bearing F9 tumors. This was due to the intratumoral administration of the ICs, but only at the cost of excessive toxicity. While single high dose (10 mg/kg) of paclitaxel conferred just a slight tumor growth retardation, the combination of low-dose paclitaxel (8.75 mg) with mIL12-F8-F8 led to a maintained CR in 2/4 mice bearing F9 tumors. Similar successful therapies reporting eradication of tumors by the combination of AAAs with AAAs/chemotherapeutic agents are listed in Table 3.55,57,58,63–66
VDAs combined with chemotherapy
Combination of VDAs which confer the best effects on bulky tumors with chemotherapeutic agents should be given in an opposite order to that of AAAs. Here, chemotherapeutic agents should be administered in advance to the chemotherapeutics. Such combination would lead to the occlusion of blood vessels, which entrap chemotherapeutic agents, and as a result increase retention of chemotherapeutics. In addition, VDA therapy following chemotherapy may avoid rapid clearance of chemotherapeutic drugs from the bloodstream; consequently, the therapeutic efficacy improves. However, the reverse order of drugs may produce similar beneficial effects. VDA therapy given before/along with chemotherapy leads to the activation/proliferation of dormant rim cells, which render them susceptible to the chemotherapeutic agents. The critical impact of drug schedule is clearly reflected in the study conducted by Martinelli et al. 71 In this study, authors examined the impact of schedule and sequence on combined therapeutic potential of a microtubule destabilizing VDA (ZD6126) and a microtubule stabilizing chemotherapeutic paclitaxel in mice bearing MDA-MB-435 breast tumor xenografts. Weekly cycles of ZD6126 (200 mg/kg intraperitoneal (i.p.)) were injected at different time points prior or after paclitaxel (10, 20, and 40 mg/kg i.v.). Protective effect of paclitaxel toward ZD6126 failed to produce any measurable benefit, when VDA administered 2 or 24 h after chemotherapy. Distancing drug administration by 72 h counteracted the opposing effects of paclitaxel and boosted the anti-tumor potential of VDA therapy. Furthermore, schedules in which VDA were given before chemotherapeutic were consistent with VDA-induced proliferation of tumor rim cells, leading to additive cytotoxicity. The most effective response was accomplished when VDA administered 72 h before paclitaxel (50% CR). A single injection of VDA followed by weekly chemotherapy yielded a similar response (57% CR). In another study, Siim et al. 70 verified the anti-tumor effects of DMXAA combined with several chemotherapeutic agents in murine mammary carcinoma xenografts. The observed additive effects in combination of VDA with different chemotherapeutics were in the order of 5-fluorouracil < (etoposide, carboplatin, cyclophosphamide, doxorubicin, cisplatin) < (docetaxel, vincristine) < paclitaxel. The interaction of DMXAA with paclitaxel was prominent and the combined therapy resulted in extension of the median tumor growth delay from 0.3 to 80 days, with 3 of 7 mice cured. This combination showed a broader timing, as paclitaxel produced similar activity when administered 4 h before to 1 h after VDA. Notably, VDA administration 4 h before paclitaxel conferred no cures. Green et al.72,73 demonstrated synergism effects associated with tumor cures, when DMXAA was combined with either paclitaxel (in non-small-cell lung carcinoma (NSCLC) lung and ovarian models) or docetaxel (in prostate model).
VDAs combined with immunotherapy
Immunotherapy is curative against small tumors in animal models of cancer, the foremost obstacle that must be addressed relates to the immune resistance of large tumors. This immune resistance could be surmounted by combining immunotherapy with a rigorous attack of VDAs on the vasculature of large tumors, leading to finely tuned immune responses, potentiating tumor ablation, and more importantly, eliciting anti-tumor immunity. 107 For this, Kanwar et al. 76 combined DMXAA and flavone acetic acid (FAA) with B7.1 (CD80) to trigger anti-tumor immunity in mice bearing large lymphoma xenografts. B7.1 is known to costimulates proliferation of T-cells through CD28 pathway. B7.1 complementary DNA (cDNA) was injected into EL-4 tumors and 24 h later followed by VDA therapy of either DMXAA or FAA. Employing this schedule, tumors were successfully removed within 2–6 weeks. CD8+ T-cells and NK cells were active in the combination setting, which resulted in heightened and sustained anti-tumor cytolytic activity and enhanced destruction of cells by inducing apoptosis. This combination therapy resulted in acquired tumor-specific immunity in cured animals, since treated mice were immune to a challenge of 1 × 107 parental lymphoma cells but not a challenge of 1 × 104 Lewis lung carcinoma cells. Moreover, adoptive transfer of 2 × 108 splenocytes from cured animals into the tumor bearing recipients prompted fast and full rejection of tumors within 3 weeks.
VDAs combined with anaerobic bacteria
Anaerobic bacteria are capable to selectively localize into the avascular necrotic regions of tumors which are normally inaccessible to intravenously administered drugs. 108 However, the low oxygen tension in this area confers these cells less susceptible to radiation and chemotherapeutic agents.109,110 Albeit, there are reports regarding successful treatment of established tumors by single-bacteria therapy,111–114 monotherapy with bacteria is limited mostly due to the concerns regarding bacteria toxicity and incomplete tumor lysis. 108 Since the hypoxia tension varies in different parts of the tumor, bacteria cannot consume all parts of the malignant tissue. For this, combination therapy with chemotherapeutic treatments (COBALT) may resolve the problem by offering synergic effects. Such combination may employ small molecules to kill cancer cells close to blood vessels, while bacteria could destroy the cells in the hypoxic niche, located far from tumor nourishing vessels. 28 Dang et al. 74 verified the therapeutic potential of COBALT in combination with VDAs in HCT116 colorectal cancer and B16 melanoma xenografts. Their rational was that VDAs can trap bacteria spores within avascular regions of tumors, buying time for the spores to germinate. Moreover, the vascular collapse would lower the oxygen tension close to the trapped bacteria and thereby increases the hypoxic region for bacterial growth in tumors. In addition, proliferating tumor rim cells could be killed upon exposure to the chemotherapeutic agent. To this end, 26 bacterial strains and 9 VDAs were screened to find the best candidates for combination therapy. Clostridium novyi-NT was found the best candidate due to its capability for extensive spreading throughout avascular regions of the tumors and for devoiding lethal toxins. Animals were given i.v. doses of 5 × 107 C. novyi-NT spores (time 0), followed by i.v. injection with D10 as VDA (0.3 mg/kg) at 24 h and i.p. injection with chemotherapeutic agent MMC (4 mg/kg) at 48 h. Full eradication of large tumors was observed in 50% of mice. Unfortunately, the striking anti-tumor effects of COBALT were attended with severe toxicity probably due to lysis syndrome. In another study, Dang et al. 75 explored the therapeutic performance of C. novyi-NT combined with VDAs (HTI-286 and vinorelbine) and vascular stabilizing agents (taxanes such as the docetaxel and MAC321). Results indicated that VDAs but not taxanes compromised the blood flow to tumors, which led to enlarging the hypoxic region, where germination C. novyi-NT spores could efficiently proceed. Combination of C. novyi-NT (a single injection) with VDAs was able to ablate various tumor xenografts without severe toxicity.
VDAs combined with radioimmunotherapy
The successful targeting of radio-labeled antibodies to tumor sites was directed to the advent of radioimmunotherapy (RIT). Beta particle isotopes, including 131I and 90Y, could emit their energy throughout a large portion of tumor. RIT is proved as a beneficial modality in cancer therapy since there is no need for drug internalization or binding to each individual cell to elicit cytotoxicity.34,115 VDA administration 48 h before RIT could enhance antibody tumor localization and retention and enable removal of any residual viable cells upon irradiation.68,69 Pedley et al. 68 explored the efficacy of RIT plus DMXAA in LS174T colorectal carcinoma xenografts. A single administration of DMXAA (27.5 mg/kg) failed to clear viable rim cells; however, when the same dosage was coupled with 18.5 MBq/50 µg 131I-labled anti-carcinoembryonic IgG, prolonged CR was accomplished in five of six mice. In another study, Pedley et al. 69 evaluated the efficacy of CA4P combined with RIT in SW1222 xenografts. Upon monotherapy, a narrow viable cells survived at the tumor edge, while in combination setting, 7.4 MBq 131I-labeled anti-carcinoembryonic antigen IgG plus a single 200 mg/kg dose of CA4P produced complete cures in five of six mice lasting for >9 months.
Conclusion and future direction
Vascular targeting approaches as single modalities or in combination with other anti-cancer strategies demonstrated beneficial effects in inhibition/eradication of established tumors in mice. The main impediment to complete cure, at least when a VTA is implicated, is the existence of viable residual cells called as tumor rim cells; because of their special characteristic, tumor rim cells are inaccessible and less susceptible to most anti-cancer drugs. In most cases, implementation of long-lasting toxic doses of anti-cancer agents is required to eradicate tumors in single modalities, and since these two key features (effective dose and high retention time) are beyond our grasp—at least for now—we have to employ complementary approaches with synergistic effects to eradicate rim cells for good. In this article, we discussed how the combination of VTAs with different approaches, including chemotherapy, radiotherapy, PDT, bacteria therapy, immunotherapy, and RIT, led to the additive effects, and with the aid of precise drug timing, sequencing, and scheduling, CRs of tumor were accomplished in a large number of treated animals. Besides, using ligand-directed VTAs, including ITs, ICs, and coaguligands, was approved as a worthy alternative approach over combination therapies that could perform curative effects as single modalities. We believe that focusing on novel strategies that specifically and effectively enable us to target tumor rim cells will enhance the prognosis of current anti-cancer strategies and may facilitate translation of beneficial effects accomplished in the labs to the patient’s bedside.
Footnotes
Acknowledgements
The authors sincerely appreciate professor Jain for his kind permission. R.J.E. and K.S. participated in writing the manuscript and N.Z. has edited the final draft.
Consent for publication
Consent for using Figure 1 with minor changes is granted by Professor Rakesh Jain.
Declaration of conflicting interest
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
We wish to thank Drug Applied Research Center, Tabriz University of Medical Sciences (IR) (5/79/4301) and Iran National Science Foundation (INSF) for financial support of this study (90007316).