
Abstract
Long non-coding RNA MEG3 has been identified as a tumor suppressor which plays important roles in tumorigenesis; however, its potential role in breast cancer has not been fully examined. Here, we showed that MEG3 was downregulated in breast cancer tissues and cell lines. Overexpression of MEG3 inhibited breast cancer cell proliferation and invasion, suggesting that MEG3 played an important role in breast cancer progression and metastasis. Moreover, MEG3 upregulation caused marked inhibition of angiogenesis-related factor expression. Conditioned medium derived from MEG3 overexpressed breast cancer cells significantly decreased the capillary tube formation of endothelial cells. Furthermore, elevated expression of MEG3 in breast cancer inhibits in vivo tumorigenesis and angiogenesis in a nude mouse xenograft model. Mechanistically, overexpression of MEG3 results in downregulation of AKT signaling, which is pivotal for breast cancer cell growth, invasion, and tumor angiogenesis. Collectively, these results suggest that MEG3 might suppress the tumor growth and angiogenesis via AKT signaling pathway and MEG3 may serve as a potential novel diagnostic and therapeutic target of breast cancer.
Introduction
Breast cancer is the most prevalent cancer among women worldwide and the second most common cause of cancer death in women. 1 Despite dramatic advances have been made in screening methods and treatments of breast cancer over the last decade, the overall survival of patients with breast cancer is still quite low due to the high rate of vital organ metastasis. Therefore, it is of great need to develop novel diagnostic and therapeutic agents to improve the treatment of breast cancer.
Long non-coding RNAs (lncRNAs) are genomic non-coding transcripts longer than 200 nucleotides which modulate gene expression at the transcription, translation, or epigenetic level. 2 Aberrant expression of lncRNAs has been reported in breast cancers.3,4 Increasing evidences have indicated that lncRNA play a critical role in tumor biology, including tumor initiation, progression, and metastasis. 5 Recent researches have demonstrated that dysfunction of lncRNAs is associated with tumor angiogenesis and may serve as predictor of poor recurrence-free survival in cancer.6,7 Tumorigenesis and tumor angiogenesis are two critical processes in the development and progression of cancer, thus a better understanding of the functions of lncRNAs may provide novel therapeutic insight for the treatment of breast cancer.
Maternally expressed gene 3 (MEG3) is an imprinted gene belonging to the imprinted DLK1-MEG3 locus located at chromosome 14q32.3 in humans. 8 MEG3 gene encodes a long non-coding RNA (lncRNA) and is expressed in a wide range of normal tissues.9,10 Previous studies have shown that MEG3 was lost in many types of cancer including glioma, 11 meningiomas, 12 gastric cancer, 13 and bladder cancer. 14 Overexpression of MEG3 has been shown to suppress tumor cell growth, migration, and invasion and promote cell apoptosis in vitro.11,15,16 Recently, it has also been showed that loss of MEG3 was associated with brain blood vessel development in MEG3 knockout mice brains 17 and downregulation of MEG3 promotes angiogenesis after ischemic stroke. 18 However, the role of MEG3 in breast cancers remains unclear.
The focus of this study is to reveal the roles of MEG3 in breast cancers and its potential mechanism. We observed that MEG3 was downregulated in breast cancer tissues and cell lines. Overexpression of MEG3 resulted in defects in breast cancer cell proliferation and invasion. Moreover, overexpression of MEG3 inhibited AKT signaling pathway and suppressed tumor growth and angiogenesis both in vitro and in vivo.
Materials and methods
Analysis of gene expression in breast cancer
Expression levels of MEG3 and vascular endothelial growth factor A (VEGFA) were assessed in a previous microarray dataset which included 165 breast cancer samples and 33 paired normal breast tissues. Briefly, raw .cel files were downloaded from NCBI GEO (GSE76250). The expression level of MEG3 and VEGFA was expressed in FPKM (fragments per kilobase of exon per million reads mapped) and calculated by TopHat and Cufflinks package.
Cell lines
MDA-MB-231 and MCF-7 human breast cancer cells were maintained in Dulbecco’s Modified Eagle’s Medium (DMEM; Invitrogen, USA) supplemented with 10% fetal bovine serum (FBS; Gibco, USA). MCF-10A cells were cultured in DMEM/Nutrient Mixture F-12 (F-12) 50/50 medium containing 5% FBS in the presence of 10 µg/mL insulin, 20 ng/mL epidermal growth factor, 100 ng/mL cholera toxin, and 0.5 µg/mL hydrocortisone. Human microvascular endothelial cells (HMEC-1) were cultured in MCDB 131 medium (Gibco) containing 10% FBS, 2 mM
Lentivirus packaging and transfection
To overexpress MEG3 in breast cancer cells, human MEG3 was cloned and inserted in the lentiviral expression vector pCDH-CMV-MCS-EF1-copRFP (System Biosciences, USA) located downstream of the cytomegalovirus promoter. Packaging of the pCDH expression constructs into pseudoviral particles was performed with the pPACKH1TM Packaging Plasmid mix (SBI) using Lipofectamine 2000 (Invitrogen) in a pseudoviral particle producer cell line (293T cells). The supernatant was harvested and viral particles were further concentrated as described previously. 19 Pseudoviral titer was finally determined by real-time polymerase chain reaction (RT-PCR).
For infection, MDA-MB-231 or MCF-7 cells were plated on a 12-well plate at 5 × 104 cells and infected with lentivirus at a multiplicity of infection (MOI) of 100 in the presence of 5 mg/mL polybrene. Lentivirus-transduced cells were selected with puromycin (1 µg/mL) to obtain stable clones, and the overexpression of MEG3 in lentivirus-transduced stable clones was verified by RT-PCR.
Cell proliferation assay
Cell proliferation analysis was performed with Cell Counting Kit-8 (CCK-8; Dojindo, Japan) according to the manual of the manufacturer. Briefly, transfected MDA-MB-231 and MCF-7 cells were plated in 96-well plates in triplicate at 2 × 103 cells each well and cultured in the growth medium. Cells were examined at 1, 2, 3, and 4 days. CCK-8 (10 µL) was added to each well at different time points. After an incubation of 2 h at 37°C, absorbance was measured at 450 nm. The optical density (OD) values of each well represented the cell proliferation.
Colony formation assay
Of each transfected cells, 500 cells were plated in six-well plate and cultured for 14 days. Then, cells were fixed and stained with methanol for 30 min, followed by 0.5% crystal violet for 20 min. Cell colonies were quantified in five different fields, and the mean value was calculated.
Cell invasion assays
Cell invasion was examined using an extracellular matrix membrane (BD Biosciences, USA). Briefly, cells were suspended in serum-free medium and placed in the top chambers, and complete medium containing 10% FBS was added to the bottom chambers. The chambers were then incubated for 24 h at 37°C with 5% CO2. After incubation, the noninvasive cells were gently removed from the top wells with a cotton-tipped swab and the chambers were fixed with methanol for 30 min. The chambers were then stained with crystal violet for another 30 min. Cells migrating into the lower chamber were photographed in five random microscopic fields (Leica, Germany), and each experiment was repeated three times. Quantification was done by measuring with microplate reader (OD = 570 nm) after being destained with glacial acetic acid.
Tube formation assay
HMEC-1 (1 × 104 cells/well) was seeded on a 96-well plate coated with 100 µL growth factor–reduced Matrigel™ (BD Biosciences) and cultured with 100 µL regular culture medium (culture medium), or control breast cancer cells conditioned medium (control medium), or MEG3 overexpressed breast cancer cells conditioned medium (MEG3 medium). After incubation at 37°C for 16 h, five randomly chosen fields were photographed using an inverted microscope (Leica). Total tube branch numbers and loops per image were automatically analyzed using WimTube software (Wimasis, Germany) by an independent observer.
Enzyme-linked immunosorbent assay
To determine the level of VEGFA secreted by MDA-MB-231 or MCF-7, 5 × 104 transfected cells were seeded onto six-well plates and cultured with regular culture medium. After incubation for 3 days, the cell supernatant was collected and centrifuged to remove cells and then stored at −80°C. Then, the protein secretion of VEGFA was analyzed using specific VEGFA enzyme-linked immunosorbent assay (ELISA) kits (Westang Bio-Tech, China) according to the manufacturer’s instructions. Normal culture medium was also used as control.
Tumor xenograft experiments
All animal procedures were approved by the Ethics Committee of Huazhong University of Science and Technology and performed according to the guidelines of the US Department of Health for Use and Care of Laboratory Animals. Stable transfected MDA-MB-231 cells were collected and suspended in 100 µL of phosphate-buffered saline (PBS) at a concentration of 3 × 107 cells/mL and then subcutaneously injected into the left armpits of BALB/c nude mice (n = 6). The mice were killed and the tumor masses were weighed 4 weeks after surgery. The tumor volume (V) was calculated by measuring the length (L) and width (W) of the tumor with calipers and using the formula V = 0.5 × (L × W2). Tumor tissues were also processed and sectioned for histological evaluation.
Immunohistochemistry
Immunohistochemistry staining of paraffin-embedded sections was performed according to the manufacturer instructions. Briefly, each slide was deparaffinized in 60°C, followed by treatment with xylene and graded alcohol. After the antigen retrieval and being blocked with 5% bovine serum albumin, tissue slides were incubated with Ki-67 (Cell Signaling Technology, Inc., USA) or CD31 (Santa Cruz Biotechnology, Inc., USA) and then visualized by standard avidin–biotinylated peroxidase complex method. Hematoxylin was used for counterstaining, and morphologic images were observed with a light microscope (Leica). To calculate microvascular density (MVD), the five random fields per section of most-vascularized areas of the tumor were selected and mean values were obtained by counting vessels. A single microvessel was defined as a discrete cluster of cells positive for CD31 staining, with no requirement for the presence of a lumen.
Quantitative RT-PCR analysis
Total RNA was isolated using the TRIzol reagent (Invitrogen, Life Technologies, USA), and 1 µg RNA from each sample was reverse-transcribed into complementary DNA (cDNA) and subjected to quantitative RT-PCR (qRT-PCR) using the SYBR Green I Kit ((Roche, Switzerland)) according to the manufacturer’s protocol. The 2−ΔΔCt method was used to calculate the relative expression of each gene relative to the amount of glyceraldehyde 3-phosphate dehydrogenase (GAPDH), and each qRT-PCR was performed in triplicate. Primers used for RT-PCR are summarized in Table 1.
Quantitative real-time polymerase chain reaction primer sequences.

VEGF: vascular endothelial growth factor; PGF: placental growth factor; bFGF: basic fibroblast growth factor; TGF: transforming growth factor; ANG1: angiopoietin-1; SDF-1: stromal cell–derived factor-1; MMP-9: matrix metallopeptidase-9; GAPDH: glyceraldehyde 3-phosphate dehydrogenase.
Western blot assay
Cells were washed with PBS and lysed in radioimmunoprecipitation assay (RIPA) buffer with phosphorylase inhibitor and protease inhibitor for 10 min on ice. Protein concentration was measured using the bicinchoninic acid (BCA) assay (Thermo Fisher Scientific, USA). Equal amounts of protein (30 µg/lane) were separated on 10% sodium dodecyl sulfate (SDS) polyacrylamide gels and transferred to polyvinylidene fluoride (PVDF) membranes. The membranes were then blocked with 5% non-fat milk and incubated with primary antibodies AKT (Cell Signaling Technology, Inc.), phosphorylated AKT (Ser473; Cell Signaling Technology, Inc.), matrix metallopeptidase (MMP)-9 (Cell Signaling Technology, Inc.), and proliferating cell nuclear antigen (PCNA) (Abcam Inc., USA) overnight. GAPDH (Cell Signaling Technology, Inc.) was used as a loading control. After being washed, the membranes were incubated with horseradish peroxidase (HRP)-conjugated secondary antibodies for 2 h at room temperature. Specific binding was detected with enhanced chemiluminescence reagents. The blots were semi-quantified by ImageJ analysis software for comparisons.
Statistical analysis
All data were presented as mean ± standard deviation (SD). Statistical analysis of the data was performed using analysis of variance (ANOVA) with a Student–Newman–Keuls (SNK) post hoc analysis. The p values <0.05 were considered statistically significant.
Results
MEG3 was decreased in breast cancer tissues and cell lines
To determine the expression of MEG3 in breast cancer, we re-analyzed publicly available dataset profiling, which included 165 breast cancer samples and 33 paired normal breast tissues. Our results revealed that the expression of MEG3 was decreased in breast cancer tissues compared with adjacent normal tissues (Figure 1(a)). We then examined the expression level of MEG3 in human epithelial cells (HMEC-1), breast epithelia cell line (MCF-10A), and breast cancer cell lines (MCF-7, MDA-MB-231). Compared to HMEC-1, MEG3 expression was significantly downregulated in cancer cell lines (Figure 1(b)).

Downregulation of MEG3 in breast cancer tissues and cell lines. (a) Analysis of MEG3 expression levels in 165 breast cancer samples and 33 paired normal breast tissues. (b) qPCR analysis of MEG3 expression levels in MDA-MB-231, MCF-7, MCF-10A, and HMEC-1 (*p < 0.05).
MEG3 overexpression impairs breast cancer cell growth and invasion
Since MEG3 was downregulated in breast cancer tissues and cell lines, we planned to generate MEG3 overexpressed breast cancer cells to observe its function. We generated MEG3-MDA-MB-231 and MEG3-MCF-7 by stable transfection of lentivirus MEG3. Quantitative polymerase chain reaction (qPCR) results confirmed that the expression levels of MEG3 in MEG3-MDA-MB-231 and MEG3-MCF-7 were upregulated ~45.9 fold and ~26.3 fold (Figure 2(a)). Compared with breast cancer cells transfected with nonspecific scramble control, CCK-8 assay showed that overexpression of MEG3 in MDA-MB-231 and MCF-7 cells significantly decreased the breast cancer cell proliferation (Figure 2(b)). In addition, colony formation assay showed that overexpression of MEG3 could remarkably inhibit breast cancer cell colony formation (Figure 2(c) and (d)). We then explored whether MEG3 regulates breast cancer cell invasion. Transwell assay showed that breast cancer cell migration was remarkably impaired in those transfected with MEG3 compared with those transfected with the control (Figure 2(e) and (f)). Taken together, these data suggested that MEG3 could regulate breast cancer cells’ growth and invasion.

MEG3 overexpression suppresses breast cancer proliferation and invasion in vitro. (a) qPCR analysis of the expression of MEG3 in control or MEG3 stably transfected MDA-MB-231 and MCF-7 cells. (b) CCK-8 analysis of the cell proliferation of control or MEG3 MDA-MB-231 and MCF-7 cells at indicated time. (c and d) Soft agar analysis shows the colony formation ability of control or MEG3 MDA-MB-231 and MCF-7 cells. (e and f) Transwell assay evaluates the effect of MEG3 overexpression on MDA-MB-231 and MCF-7 cells’ invasive capacity (bars = 100 µm; *p < 0.05).
MEG3 overexpression suppresses tumor angiogenesis in vitro
Angiogenesis plays an important role in the growth and metastasis of breast cancer; hence, the change in the expression of angiogenesis-related genes (VEGFA, vascular endothelial growth factor B (VEGFB), placental growth factor (PGF), basic fibroblast growth factor (bFGF), stromal cell–derived factor 1 (SDF-1), transforming growth factor (TGF)-β1, Angiogenin, and MMP-9) was evaluated between control and MEG3 overexpressed breast cancer cells. As shown in Figure 3(a) and (c), the messenger RNA (mRNA) levels of VEGFA, PGF, bFGF, TGF-β1, and MMP-9 were significantly decreased when MEG3 was overexpressed in MDA-MB-231 and MCF-7 breast cancer cells. The consistent change in VEGFA protein secreted into the culture medium was observed in breast cancer cells transfected with MEG3 (Figure 3(b) and (d)). Meanwhile, VEGFA was significantly overexpressed in breast cancer tissues in comparison with paired normal tissues (Figure 3(e)). Interestingly, we evaluated whether VEGFA expression was associated with the expression of MEG3 and it turned out that VEGFA expression was significantly negatively correlated with MEG3 expression by analyzing the above 165 breast cancer tissues (Figure 3(f), R = −0.4683, p < 0.0001).

Overexpression of MEG3 suppresses angiogenesis-related molecule expression in breast cancer. (a) qPCR analysis of the expression of angiogenesis-related genes in control or MEG3 MDA-MB-231 cells. (b) ELISA analysis of the expression of VEGFA protein in control or MEG3 MDA-MB-231 cells culture media. (c) qPCR analysis of the expression of angiogenesis-related genes in control or MEG3 MCF-7 cells. (d) ELISA analysis of the expression of VEGFA protein in control or MEG3 MCF-7 cell culture media. (e) Expression of VEGFA mRNA level in 165 breast cancer samples and 33 paired normal breast tissues (*p < 0.05). (f) VEGFA mRNA level was inversely correlated with MEG3 level in breast cancer tissues (Spearman’s correlation analysis, r = −0.4683, p < 0.0001).
To further confirm the role of MEG3 in tumor angiogenesis, an in vitro capillary tube-formation assay was performed. As shown in Figure 4, the capillary tube formation of HMEC-1 cultured in control-MDA-MB-231- and control-MCF-7-derived conditioned medium was significantly increased compared with those cultured in culture medium. HMEC-1 grown in MEG3-MDA-MB-231- and MEG3-MCF-7-derived medium formed less capillary branches and tubes than those grown in control-MDA-MB-231- and control-MCF-7-derived medium. Taken together, these results suggest that MEG3 lost in breast cancer cells may promote angiogenesis-related gene such as VEGFA expression and thus stimulate tumor angiogenesis.

MEG3 overexpression inhibits tumor angiogenesis in vitro. (a) Representative tube formation images of HMEC-1 cultured with control or MEG3 MDA-MB-231- and MCF-7-cell-derived conditioned medium (bars = 100 µm). (b and c) Bar graph shows the total branching points and loops of tube formation (*p < 0.05).
MEG3 overexpression suppresses tumorigenesis and angiogenesis in vivo
Next, we further determined whether elevated expression of MEG3 in breast cancer cells influences tumorigenesis and angiogenesis in vivo. Control-MDA-MB-231 or MEG3-MDA-MB-231 cells were subcutaneously injected to establish a nude mouse xenograft model. Tumor volumes and weights in mice injected with MEG3-MDA-MB-231 cells were significantly smaller than those injected with control-MDA-MB-231 cells (Figure 5(a)–(c)). Ki-67 is widely used as a “proliferation marker” to measure the growth fraction of cells in tumors. To further confirm that MEG3 inhibits proliferation of xenograft tumors, we performed immunohistochemical staining of Ki-67. As is shown in Figure 6(a) and (b), expression of Ki-67 was downregulated in tumors from mice injected with MEG3-MDA-MB-231 cells when compared with the control, indicating that MEG3 can inhibit proliferation of breast cancer cells in vivo. Additionally, CD31 immunostaining showed a significantly decreased MVD in tumors from MEG3-MDA-MB-231-injected mice compared with those from control-MDA-MB-231-injected mice (Figure 6(a) and (c)). Collectively, these data demonstrate that MEG3 suppresses tumorigenesis and the formation of new blood vessels in vivo.

MEG3 overexpression suppressed tumorigenicity in vivo. (a) Control or MEG3 MDA-MB-231 cells were subcutaneously injected into the nude mice (n = 6). Tumors were harvested 28 days later and photographed. (b) Growth curve of control and MEG3 MDA-MB-231 cells’ xenograft tumors was drawn by measuring tumor volumes at the indicated days. (c) Bar graph shows the weight of control and MEG3 MDA-MB-231 cells’ xenograft tumors (*p < 0.05).

Elevated expression of MEG3 suppresses tumor growth and angiogenesis in a nude mouse xenograft model. (a) Representative image shows the expression of Ki-67 and CD31 in breast cancer xenografts (bars = 50 µm). (b) Bar graph shows the percentage of Ki-67-positive cells in control and MEG3 MDA-MB-231 cells’ xenograft tumors. (c) Bar graph shows the MVD in control and MEG3 MDA-MB-231 cells’ xenograft tumors (*p < 0.05).
MEG3 overexpression suppresses AKT pathways in breast cancer
AKT pathway is often aberrantly activated and contributes to cell proliferation, migration, and tumor angiogenesis in human breast cancers.20,21 To determine the possible mechanism by which MEG3 regulated proliferation, migration, and tumor angiogenesis of breast cancer cells, western blot analysis was performed to investigate the effects of MEG3 overexpression on AKT signaling pathways. As a result, western blot analysis showed that MEG3 overexpression significantly reduced the levels of phosphorylated AKT, while no detectable changes were observed in the total AKT (Figure 7(a) and (b)). In addition, PCNA, MMP-9, and VEGFA, which are known as downstream factors of AKT signaling in regulating tumor proliferation, metastasis, and angiogenesis, were downregulated after MEG3 overexpression. These results indicated that MEG3 overexpression might suppress breast cancer cell proliferation, migration, and tumor angiogenesis through AKT signaling.
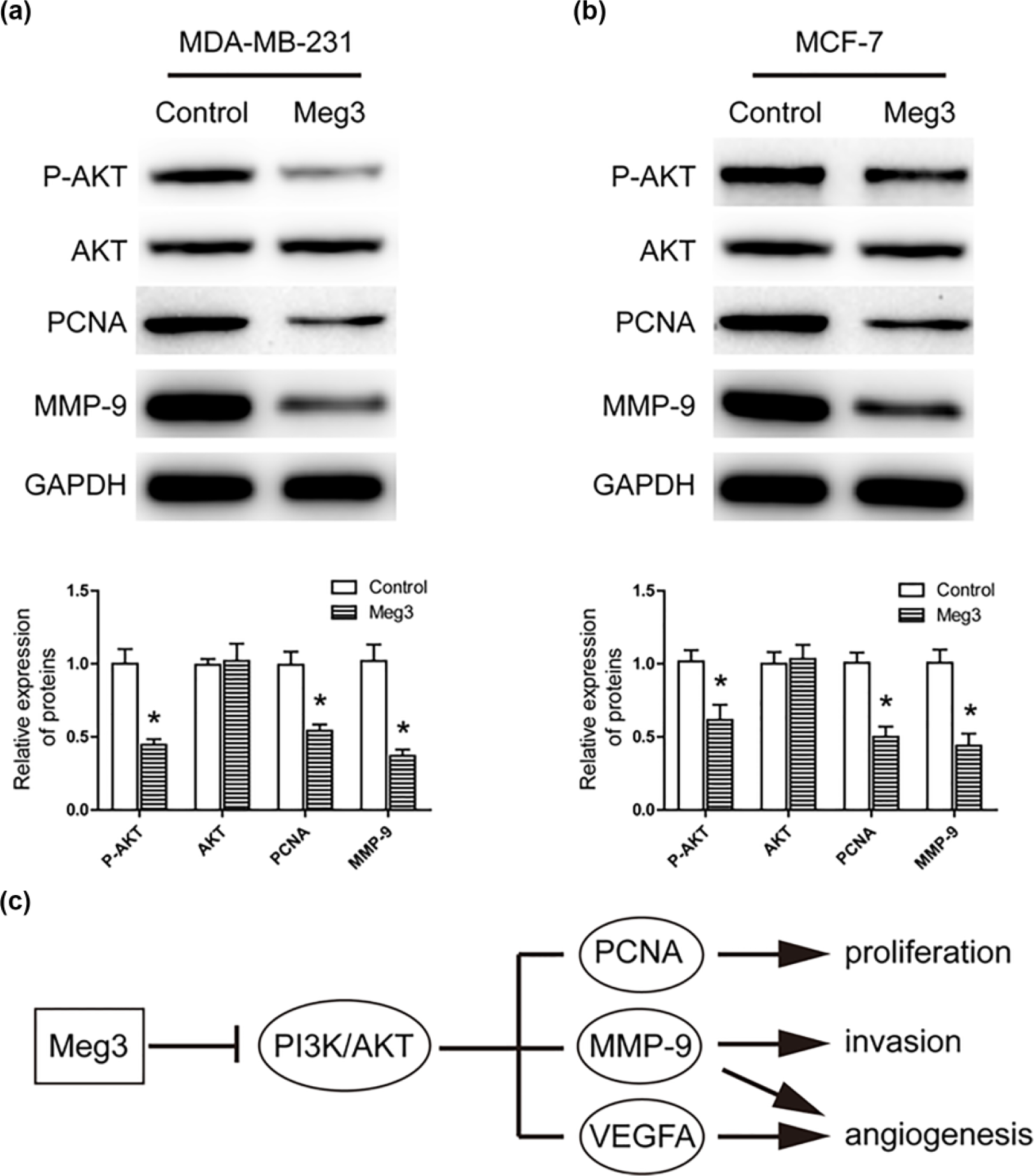
Overexpression of MEG3 inhibits AKT pathway signaling in breast cancer cells. (a) Western blotting analysis of the expression of AKT, p-AKT, PCNA, and MMP-9 in control or MEG3 MDA-MB-231 cells. (b) Western blotting analysis of the expression of AKT, p-AKT, PCNA, and MMP-9 in control or MEG3 MCF-7 cells. (c) A proposed model for MEG3 overexpression regulated AKT pathway and subsequent cell proliferation, invasion, and tumor angiogenesis in breast cancer.
Discussion
During recent years, the role of lncRNAs in tumorigenesis gets more and more attention, such as HOX antisense intergenic RNA (HOTAIR), metastasis-associated lung adenocarcinoma transcript 1 (MALAT1), lncRNA associated with microvascular invasion in hepatocellular carcinoma (HCC; MVIH), and MEG3. Accumulating evidences suggested that these lncRNAs are involved in the biological behavior of cancer cells, including proliferative capability and replicative immortality, activating invasion and metastasis, inducing angiogenesis, and resisting cell death.6,22–24 In this study, we investigate the role and underlying mechanisms of lncRNA MEG3 in breast cancers. Our findings show that ectopic expression of MEG3 suppresses AKT signaling pathway and thereby regulates breast cancer cell biology and tumor angiogenesis in vitro and in vivo.
MEG3 was initially discovered as a tumor-associated lncRNA and its expression was downregulated in many tumors, such as lung cancer, 15 nasopharyngeal carcinoma, 25 meningioma, 12 glioma, 11 and gastric cancer. 13 Recent researches show that MEG3 plays a negative role in tumor cell proliferation, apoptosis, migration, invasion, or the metastatic spread of tumor cells, indicating MEG3 as a tumor suppressor in human cancer. A very recent study showed that MEG3 was downregulated in breast cancer tissues. 26 Sun also found that the expression of MEG3 was decreased in breast cancer despite of their subtypes. 16 Consistent with these reports, our results showed that MEG3 was downexpressed in breast cancer tissue samples and breast cancer cell lines. To further clarify the biological functions of MEG3 in breast cancer, we evaluated the effect of MEG3 overexpression on breast cancer cell proliferation and invasive potential by CCK-8, colony formation, and Transwell assays. As a result, overexpression of MEG3 significantly suppressed cell proliferation and invasion in MDA-MB-231 and MCF-7 cell lines. In addition, tumor xenograft experiments revealed that overexpression of MEG3 significantly reduced the capability of breast cancer cells to induce tumorigenesis. These data indicate that MEG3 serves as a tumor suppressor in breast cancer.
Angiogenesis is essential for development and progression of breast cancer. 27 VEGFA, TGF-β1, PGF, bFGF, and MMP-9 are important mediators of tumor angiogenesis in various cancers. In this study, we found that overexpression of MEG3 in breast cancer cells led to a decrease in the expression of VEGFA, TGF-β1, PGF, bFGF, and MMP-9 in MEG3 overexpressed breast cancer cells. Previous studies showed that MEG3 was inversely associated with VEGFA levels in osteoarthritis 28 and MEG3 regulated TGF-β expression in breast cancer cells. 29 In line with previous results, our results showed that the secretion of VEGFA was decreased in MEG3 overexpressed breast cancer cells culture medium. Moreover, the expression of VEGFA was negatively correlated with MEG3 in breast cancer tissues. In addition, culture medium generated from MEG3 overexpressed breast cancer cells suppressed the capillary tube formation of endothelial cells, an important step of tumor angiogenesis. Consistently, tumors formed by MDA-MB-231 cells with upregulated MEG3 showed few microvessels in the nude mouse models, compared to controls. These results suggest that lost expression of MEG in breast cancer tissues may account for the secretion of angiogenesis factors such as VEGFA and, in turn, activates angiogenesis as well as the accelerated growth and metastasis of breast cancer.
The aberrantly activated AKT signaling is critical in the progression of breast cancer by promoting cell growth and tumor angiogenesis.20,21 Previous studies have shown that disregulated lncRNA is tightly related with phosphoinositide 3-kinase (PI3K)/AKT signaling.30–32 In this study, our data showed that MEG3 overexpression had no effect on the amount of total AKT, but decreased phosphorylated levels of AKT. Furthermore, PCNA, 33 MMP-9, 34 and VEGFA 35 are downstream targets of PI3K/AKT signaling, which are related to tumor proliferation, metastasis, and angiogenesis in mammary carcinoma. We found that MEG3 overexpression suppressed cell growth, invasion, and angiogenesis through downregulation of PCNA, MMP-9, and VEGFA expression. In agreement with our study, Qiu et al. 36 showed that MEG3 regulated PI3K/AKT signaling pathway in diabetic retinopathy. These findings suggested that MEG3 might function as a tumor suppressor in breast cancer via inhibition of the AKT signaling (Figure 7(c)).
Conclusion
In summary, this study demonstrates that MEG3 is downregulated in breast cancer. MEG3 overexpression inhibits tumor growth and angiogenesis in breast cancer through AKT pathway. Considering the anti-tumor and anti-angiogenesis properties of MEG3, MEG3 may be a potential therapeutic target for breast cancer.
Footnotes
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was financially supported by the National Natural Science Foundation of China (No. 81172512).