
Abstract
Esophageal squamous cell carcinoma is one of the deadliest of all the cancers. Its metastatic properties portend poor prognosis and high rate of recurrence. A more advanced method to identify new molecular biomarkers predicting disease prognosis can be whole exome sequencing. Here, we report the most effective genetic variants of the Notch signaling pathway in esophageal squamous cell carcinoma susceptibility by whole exome sequencing. We analyzed nine probands in unrelated familial esophageal squamous cell carcinoma pedigrees to identify candidate genes. Genomic DNA was extracted and whole exome sequencing performed to generate information about genetic variants in the coding regions. Bioinformatics software applications were utilized to exploit statistical algorithms to demonstrate protein structure and variants conservation. Polymorphic regions were excluded by false-positive investigations. Gene–gene interactions were analyzed for Notch signaling pathway candidates. We identified novel and damaging variants of the Notch signaling pathway through extensive pathway-oriented filtering and functional predictions, which led to the study of 27 candidate novel mutations in all nine patients. Detection of the trinucleotide repeat containing 6B gene mutation (a slice site alteration) in five of the nine probands, but not in any of the healthy samples, suggested that it may be a susceptibility factor for familial esophageal squamous cell carcinoma. Noticeably, 8 of 27 novel candidate gene mutations (e.g. epidermal growth factor, signal transducer and activator of transcription 3, MET) act in a cascade leading to cell survival and proliferation. Our results suggest that the trinucleotide repeat containing 6B mutation may be a candidate predisposing gene in esophageal squamous cell carcinoma. In addition, some of the Notch signaling pathway genetic mutations may act as key contributors to esophageal squamous cell carcinoma.
Keywords
Introduction
Esophageal squamous cell carcinoma (ESCC) is the eighth most common cancer, with more than 480,000 new cases and 400,000 deaths each year worldwide. 1 One of the world’s highest rates of ESCC is found in northeastern Iran, which lies in the esophageal cancer belt that stretches from China to northeastern Iran. 2 The reasons for such high rates of ESCC occurrence in this area are not fully clear. ESCC is the second leading cause of cancer-related deaths in northeastern Iran. Statistical data have demonstrated that its rates with age standardized in males and females are (5.6–10.30)/105 and (5.7–19.8)/105, respectively.2,3 Despite advances in multimodality therapy, the aggressiveness of human squamous cell carcinomas (SCCs) caused poor prognosis with average 5-year survival rates <10%. SCCs are usually diagnosed in the advanced stages and respond poorly to chemotherapy; thus, novel therapeutic strategies in the treatment of this disease are required.4,5 ESCC is a multifactorial disease; in addition to environmental factors, genetic variations contribute to its susceptibility risk. Better understanding of the molecular mechanisms underlying esophageal carcinogenesis will lead to the development of new diagnostic technologies, treatment modalities, and preventive approaches. 5
For the past several decades, the genes underlying Mendelian disorders have been identified through physical mapping, positional cloning, candidate gene sequencing, and a process of meiotic mapping. 6 Recently, the utilization of next-generation sequencing (NGS) technologies has facilitated interrogation of the genomes of human cancers. 7 Whole exome sequencing (WES) allows a targeted enrichment and resequencing of nearly all exons of protein-coding genes. Accordingly, causative mutations in Mendelian disorders have been provided by WES using next generation technologies. 8
The Notch signaling pathway is crucial for the development and homeostasis of most tissues and is evolutionarily conserved. It plays critical roles in balancing cell survival, proliferation, differentiation, and apoptosis, as well as maintenance of progenitor stem cell populations. Aberrant activation of Notch signaling has been revealed during the carcinogenesis of a variety of human cancers.9,10
Clinical trials of Notch signaling pathway inhibitors have been reported in patients with solid tumors, including breast cancer and T-cell acute lymphoblastic leukemia (T-ALL), 10 and preclinical evaluation of several approaches is under consideration. 11 However, the roles of the Notch signaling pathway in esophageal tumor biology remain elusive. 12 Because of its main role in triggering T-ALL, Notch is often considered as a model hematopoietic proto-oncogene. 13
It was shown that the risk of developing esophageal cancer is greater in individuals with first degree relatives with ESCC than in the general population. To be more specific, offspring of two parents with ESCC has an eight-fold greater likelihood of developing ESCC than offspring of parents without ESCC. 14
Currently, genetic analyses of diseases, such as cancer, have focused on the detection of rare variants that may contribute to individual susceptibility to common complex diseases. 15 It is a matter of debate whether several DNA sequence variants with high penetrance are the major contributors to disease. Based on the common disease–rare variant (CDRV) hypothesis, we aimed to find novel mutations in ESCC that can be introduced as biomarkers to be used for early diagnosis, prognosis, and possible novel therapeutics. 16
Materials and methods
Patients
Nine ESCC probands from nine unrelated families participated in this study. All samples came from individuals residing in Khorasan or Golestan provinces in northeastern Iran, located in the ESCC belt. The patients’ age ranged from 30–73 years. They were histologically confirmed to have ESCC at Omid or Emam Reza Hospitals between 2009 and 2011. All the patients had at least two consecutive generations of familial cancer or two or more affected family members in a generation. After enrollment, patients’ clinical information was collected and genomic DNA was extracted from peripheral blood, followed by exome sequencing.
Ethical approval
All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards.
Informed consent
Informed consent was obtained from all individual participants included in this study.
Exome sequencing
DNA sample concentrations were 50–300 ng/µL (measured by NanoDrop, A260/280 = 1.7–2), and 3–5 µg of DNA was utilized per reaction (Thermo Fisher Scientific, Wilmington, USA). Covaris S2 equipment was applied to shear the DNA, resulting in an optimal range of fragment-size distribution. Library size and concentration were assessed by capillary electrophoresis (Bioanalyzer; Agilent, Santa Clara, CA). The SureSelect Target Enrichment System was utilized for post-capture pooling of enriched sequencing-ready libraries. The whole exome was characterized using the Illumina Genome Analyzer II (Illumina, San Diego, CA). Mapping generated reads against the human reference genome University of California, Santa Cruz (UCSC) hg19 were obtained with the Burrows-Wheeler Alignment (BWA) tool alignment algorithm, which efficiently aligned short sequence reads against a large reference sequence (rs). Reads were processed by SAMtools to detect single-nucleotide polymorphisms (SNPs) and insertions/deletions (indels) and to remove polymerase chain reaction (PCR) duplicates (http://samtools.sourceforge.net).
Data analysis
Real-Time Analysis (RTA) software (RTA V.1.12-4) was started on-instrument, which is integrated with the Genome Analyzer Sequencing Control software (SCS). It is followed by off-instrument data analysis with Illumina Consensus Assessment of Sequence and Variation (CASAVA) software (V1.8.2). Genes associated with the Notch signaling pathway were selected according to GeneCards (http://www.genecards.org/) software. Variants with total depth (>15) and those with allelic frequencies ≥0.01% were filtered based on the data available in the 1000 Genomes Project (http://www.1000genomes.org) and dbSNP (http://www.ncbi.nlm.nih.gov/SNP/). Only variants in exonic region revealing indels, non-synonymous, and stop-gain changes were selected. Deleterious amino acid changes were selected with PolyPhen (score >0.85; http://genetics.bwh.harvard.edu/pph2) and Sorting Intolerant from Tolerant (SIFT; score <0.05; http://sift.jcvi.org/). Only non-synonymous amino acid changes were selected. Indels, which illustrate damaging deletions, were identified with MutationTaster (score >0.5; http://www.mutationtaster.org) and Protein Variation Effect Analyzer (PROVEAN; http://provean.jcvi.org/index.php). Moreover, the functional impact of amino acid substitutions in proteins was predicted with MutationTaster. Terms and previous bibliographies of variants according to National Center for Biotechnology Information (NCBI) Gene and Reactome (http://www.reactome.org/PathwayBrowser/) were evaluated for biological functions and pathways of the gene products.
The initial results may have contained false-positive reports, which were omitted using the method by Fuentes Fajardo et al. 17 and Esteban-Jurado et al. 18 Results were compared with the data of 51 healthy individuals (100 alleles) to eliminate possible polymorphisms.
To generate exact data, novel mutations from SIFT and 1000 Genomes Project were analyzed with the Exome Aggregation Consortium (ExAC) database, and mutations with allelic frequencies under 1 × 10−4 were selected (http://exac.broadinstitute.org/). GNCPro (http://gncpro.sabiosciences.com/), STRING (http://string-db.org/), and GeneMANIA (http://www.genemania.org/) software were used to demonstrate gene–gene interactions and explain the effects of other genes on the selected candidate mutations.
The miRBase database is utilized to search published micro RNA (miRNA) sequences and annotations to predict hairpins in the miRNA transcripts with information on the location and sequence of the mature miRNA sequence (www.mirbase.org).
After detection of a candidate gene in family 93 (CD3D), molecular analyses was performed to determine its familial variation. Primers for mutation detection with amplification-refractory mutation system–PCR (ARMS-PCR) were designed using Primer3web (http://primer3.ut.ee/), and checked with AlleleID and Gene Runner software applications. To confirm mutation detection and WES results, samples were sequenced using Sanger technique. Molecular analysis of CD3D in family 93 was used to evaluate genetic segregation.
Results
Whole exomes from nine probands with familial ESCC from nine unrelated families were sequenced. Clinical features of the ESCC patients are shown in Table 1. Pedigrees of six of the ESCC patients are shown in Figure 2. Of note, patients 68-1, 149-1, and 150-1 consumed hot tea, which is a known risk factor for ESCC.
Clinical features and cancer history of patients.
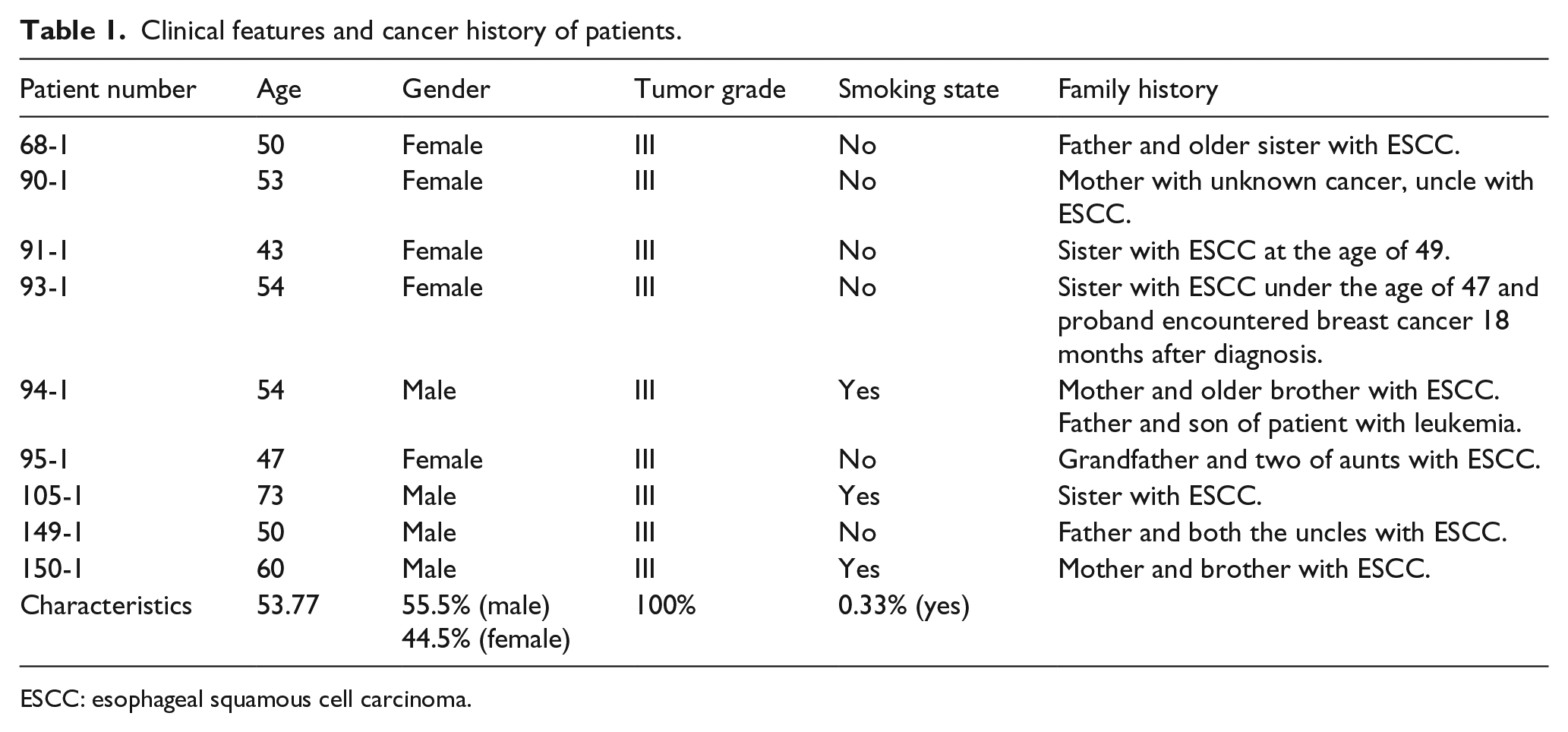
ESCC: esophageal squamous cell carcinoma.
After exome sequencing, about 600,000 variants in all cases were detected where only exonic mutations were filtered in the following steps. All levels of filtering are depicted in Figure 1. Notch signaling pathway genes were selected from GeneCards and compared with our data to extract non-synonymous, indels, and stop-gain variants. Moreover, subjects were filtered a second time to remove variants with total depth greater than 15 and allelic frequency less than 0.01, which found 232 mutations in the nine probands. Variants were detected as damaging if reported in PolyPhen and SIFT/PROVEAN for point mutations and PROVEAN and MutationTaster for indels.

Schematic data analysis after whole exome sequencing. Nine patients from different families were sequenced for familial ESCC.
Manual inspection of 68 variants resulted in no false positives among our data. We found 45 mutations that did not have rs numbers in 1000 Genomes Project or were novel in SIFT. After all filtration procedures, final results were reached by analyzing with ExAC, which eliminated another 18 more mutations, leaving 27 mutations as exonic, novel, low allelic frequency, damaging, total depth >15, and without rs numbers. Comparing these results with 51 normal genome sequences further confirmed that these 27 mutations are perhaps novel mutations and not polymorphisms. The final prioritized variants are shown in Supplementary 1.
All the mutations reported by WES were checked on the Notch signaling pathway to demonstrate the fundamental genes, with relevance scores of 10–14 in GeneCards (http://www.genecards.org/), in this pathway. Among the main genes in this category four of nine mutations were revealed as damaging, indicating a need for further analysis of Notch signaling pathway genes.
Gene–gene interaction
Four candidate gene products, epidermal growth factor (EGF), MET, AKT1, and growth differentiation factor 15 (GDF15) act through a cascade that downregulates GDF15 (growth factor) gene expression. All these genes are proto-oncogenes, and their genetic variations may cause cancer susceptibility. Detailed data on the distribution of mutated genes in each family are shown in Figure 2. Familial ESCC criteria were confirmed in all pedigrees.

Pedigrees from families 91, 94, 149, 150, 93, and 95 showing the key Notch signaling pathway mutations in probands of each pedigree.
Although these variants appeared in different pedigrees, only EGF and AKT1 were altered in the same proband (149-1). Moreover, signal transducer and activator of transcription 3 (STAT3) mutated in proband 91 is upregulated by EGF (mutated in proband 149) and downregulated by MET (mutated in proband 150) (http://gncpro.sabiosciences.com/). Detailed interactions among candidate genes are depicted in Figure 3. In conclusion, in this study the end results of all mutations found in various genes in the Notch pathway resulted in downregulation of GDF15 gene. Three other co-expressed candidate genes were CD3D in family 93, CD3E in family 94, and LCK in family 95.
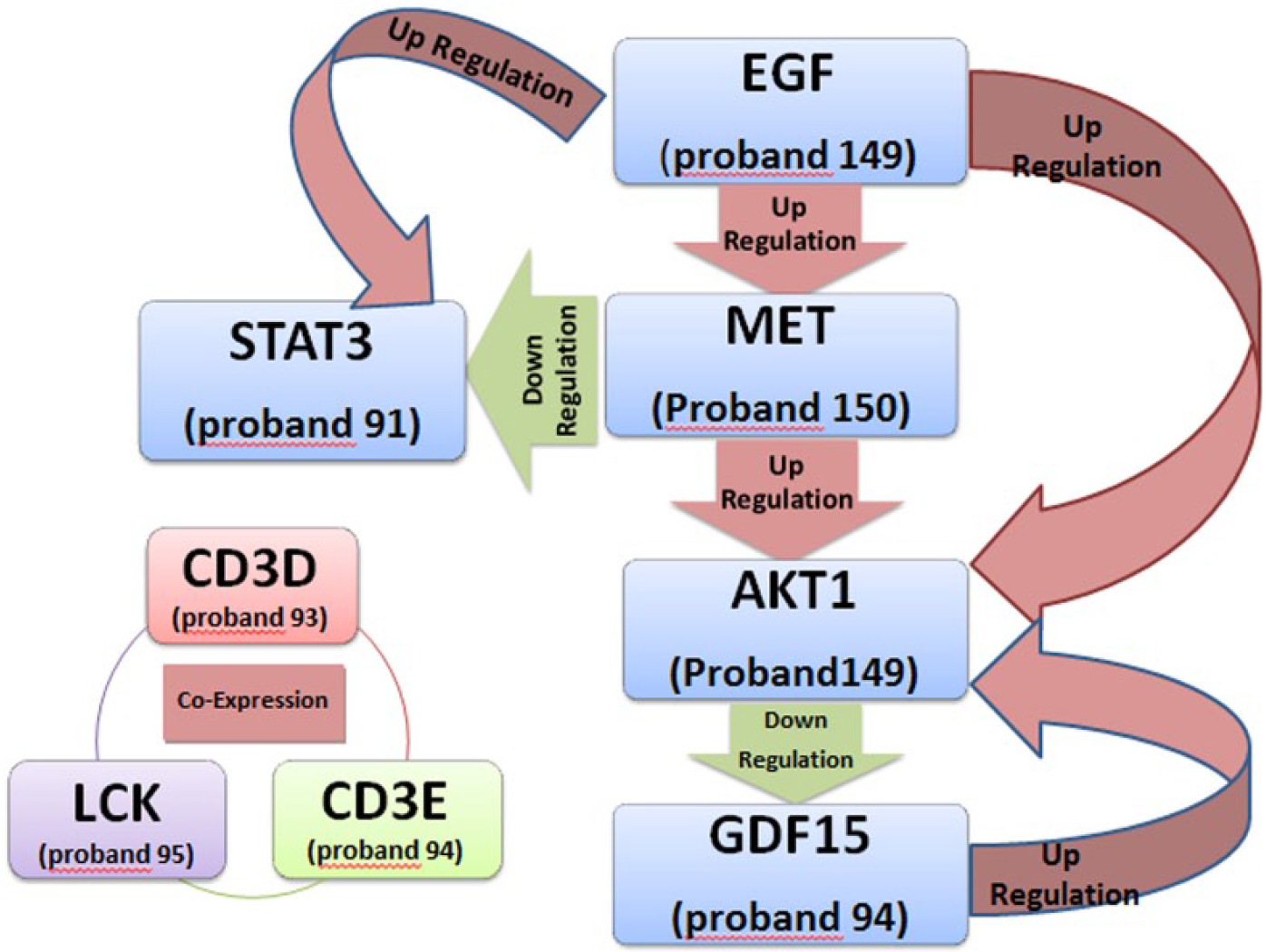
Gene–gene interactions among ESCC candidate genes.
Segregation study
Among these variants, the CD3D gene was analyzed in two individuals in family 93. The observed mutation resulted in a serine to cysteine substitution in cytoplasmic domain of the protein, which is involved in T-cell development and signal transduction. It was demonstrated that this mutation segregates through the generations (pedigree 93).
This mutation was transferred to three of the five healthy children of the proband. Neither the proband’s affected sister nor did her children inherited this mutation (Figure 4). The proband developed a primary breast tumor 2 years after she was first diagnosed with ESCC.

CD3D gene mutation pattern in family 93((+/−) heterozygote, (−/−) homozygote wild).
Trinucleotide repeat containing 6B
After various filtration procedures, trinucleotide repeat containing 6B (TNRC6B) gene variation was detected in five of nine probands. This mutation has a rs number; however, it was not found on the ExAC database. The mutation was a deleterious CAG repeat deletion found in probands 68-1, 93-1, 105-1, 149-1, and 94-1. Of cases with mutations, only sample 94-1 had six-nucleotide deletion, two repeats of CAG; however, others had three-nucleotide deletions, only one repeat unit. This variant results in a polyglutamine and alters the length of the amino acid repeat. Amino acid sequence analysis of mutated TNRC6B demonstrated that protein features might be affected by alterations in the splice site. These alterations change the donor sequence of the splice site in this region and increase the rate of splicing (MutationTaster data: wild type = 0.33/mutant = 1).
Discussion
Analysis of candidate mutations found that all the selected genes were proto-oncogenes. All the different mutations in the families belong to the Notch signaling pathway, which is involved in cell proliferation and survival. Previous studies of genetic variations revealed that many genes in this pathway predispose patients to various cancers, especially ESCC. Germ-line defects of Checkpoint kinase 2 (CHEK2) predisposed patients to familial breast cancer 19 and mutations in NOTCH1, NOTCH2, and NOTCH3, other Notch pathway components contributed to ESCC susceptibility. 20
EGF, the first gene in the Figure 3 cascade, is a potent mitogenic factor encoded by multiple transcript variants that result from alternate splicing. The EGF mutation found in our WES filtering strategy was an A46C mutation located in exon 1. This change causes a non-synonymous variation in amino acids S16R, and bioinformatics analysis confirms its damaging effect. This finding fits the fact that many of the EGF receptor family members are aberrantly altered in human carcinomas. 21
The next gene of the cascade, MET, is a receptor tyrosine kinase hepatocyte growth factor receptor that binds to its ligand and triggers downstream reactions, including proliferation, scattering, morphogenesis, and survival (UniProtKB: 08581 (MET_HUMAN)). TheA462G mutation of this gene in exon 2 resulted in an I154M change in which polar isoleucine is replaced with non-polar methionine. This polarity change may alter protein function and cause uncontrolled cell proliferation. In addition, this mutation may affect the functional domain of the polypeptide.
STAT3, which is influenced by EGF and MET, acts as a signal transducer by binding with DNA and upregulating the expression of various genes involved in cellular proliferation, migration, invasion, and angiogenesis, and is a hot spot for cancer initiation. 22 Mutation of G1621A, in STAT3 exon 18, replaces non-polar glycine with positively charged arginine at residue 541 of the protein.
AKT1, downstream of MET, EGF, and GDF15, is related to the family of serine/threonine protein kinases AKT1, AKT2, and AKT3. AKT kinases are activated by a number of growth factors and cytokines. 23 It has been indicated that the AKT1 isoform has a more specific role than the others in cell motility, proliferation, differentiation, growth, and plasticity, and is an integral node in multiple signaling pathways. 24 The AKT1 exon 10 mutation of C727T, causes amino acid change from non-charged to positively charged amino acid in the protein kinase domain of the product. This amino acid change may contribute to protein function in cancer formation.
Expression of GDF 15, the last gene in this cascade is low under normal conditions; however, it is greatly upregulated during the inflammation response. Moreover, GDF15 is a downstream target of p53 that may be involved in cancer. The G473T mutation in exon 2 of GDF15 results in replacing a positively charged arginine with a non-polar aliphatic leucine. Malfunction of GDF15 may be the result of the alteration in protein charge.
TNRC6B has been shown to stably associate with Argonaute proteins to work in an effector step in RNA interference (RNA mediated gene silencing). 25 It was confirmed that exonic variation of TNRC6B is associated with progression of myelodysplastic syndrome to acute myeloid leukemia. 26 Although it has been confirmed that TNRC6B variants contribute to tumorigenesis of different cancers such as prostate, lung SCC, and lung adenocarcinoma through downregulation of TNRC6B, it is still under debate that it could be a diagnostic marker in other cancers such as ESCC.27–29 Studies with hepatoma cell lines revealed that carcinogenesis of TNRC6B through its downregulation is due to accelerated cell proliferation and loss of cell adhesion ability. 30 TNRC6 family genes have shown mutations in five different pedigrees and based on its analysis in 51 healthy samples, it was chosen as a novel damaging mutation with a possible predisposition to ESCC. This mutation occurred as a three-nucleotide repeat; hence, its deletion did not result in a frame shift; however, this change can alter splice sites, which may affect gene expression.
miRNAs—hsa-miR-2467-3p, hsa-miR-324-3p, and hsa-miR-1538—bind to the denoted region of deletion mutation. Our findings suggest that the TNRC6B mutation may cause difficulties in miRNA binding to its messenger RNA (mRNA); so, it does not undergo miRNA-mediated decay and causes extensive cell proliferation due to its overexpression (www.mirbase.org). Further studies are needed to confirm this phenomenon. It is predicted that this deletion may cause changes in histone modification sites, open chromatin, or transcription factor binding sites such as the histone H3 lysine 36 tri-methylation and Pax5 transcription factor binding site.
The CAG deletion mutation in TNRC6B exists in the RNA recognition motif in the silencing domain and interacts with the CNOT1 and PAN3 regions of the protein, which may inhibit protein interaction with the miRNAs (UniProtKB: Q9UPQ9 (TNR6B_HUMAN)).
Previous studies revealed that the expression of the immune-related gene, CD3D, was associated with overall survival in breast cancer patients. 3 The CD3D genetic variant as an appropriate ESCC candidate was studied in family 93 based on the online databases. It demonstrated genetic segregation among the family.
This observation suggests that the CD3D mutation may be an important genetic factor for new primary breast tumors in ESCC patients and confirms the prognostic significance of CD3D gene expression in breast cancer. 31 CD3D was shown to form a complex with many genes, including the cancer-related genes IKZF1 and TRA@, and activates modification of complexes in the cancer-related genes interleukin 2 (IL2), tyrosine-protein phosphatase non-receptor type 11 (PTPN11), and casitas B-lineage lymphoma (CBL). 32
In conclusion, exome sequencing strategies should be carefully selected and modified depending on what the data set allows. 33 Candidates for implication in ESCC predisposition have been identified after rigorous scrutiny and filteration from the final ESCC variant list. This study identified several genes that may be associated with susceptibility to ESCC, including TNRC6B, CD3D, CD3E, LCK, STAT3, MET, EGF, AKT1, and GDF15. Furthermore, to enhance the efficiency of whole exome studies, improved methods, such as utilizing a provisional gene exclusion list, must be implemented.
Footnotes
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.