
Abstract
Chimeric antigen receptor modified T cell–based immunotherapy is revolutionizing the field of cancer treatment. However, its potential in treating bile duct carcinoma has not been fully explored. Herein, we developed the second-generation mesothelin-targeting chimeric antigen receptor–modified T cells with the 4-1BB co-stimulatory module by the piggyBac transposon system. Mesothelin-targeting chimeric antigen receptor was expressed by 66.0% of mesothelin-targeting chimeric antigen receptor–modified T cells post electrophoretic transfection and stimulation with K562-meso cells; the expressions of activation markers were tested by flow cytometry assay and showed greater activation of mesothelin-targeting chimeric antigen receptor–modified T cells than control T cells (CD107α: 71.9% vs 48.6%; CD27: 92.1% vs 61.8%; CD137: 55.5% vs 8.4%; CD28: 98.0% vs 82.1%; CD134: 37.5% vs 10.4%). Furthermore, mesothelin-targeting chimeric antigen receptor–modified T cells exerted cytotoxicity toward mesothelin-expressing EH-CA1b and EH-CA1a cells in an effector-to-target ratio-dependent manner, while leaving mesothelin-negative GSC-SD and EH-GB1 cells and normal liver L02 cells almost unharmed. Mesothelin-targeting chimeric antigen receptor–modified T cells secreted cytokines at higher levels when co-cultured with mesothelin-positive EH-CA1a and EH-CA1b cells than with mesothelin-negative GSC-SD and EH-GB1 cells. Enhanced cytotoxicity and cytokine secretion of mesothelin-targeting chimeric antigen receptor–modified T cells compared to control T cells were also observed when co-cultured with 293-meso cells (interferon γ: 85.1% ± 1.47% vs 8.3% ± 2.50%, p = 0.000; tumor necrosis factor α: 90.9% ± 4.67% vs 18.5% ± 3.62%, p = 0.0004; interleukin 2: 60.8% ± 2.00% vs 15.6% ± 2.06%, p = 0.002; interleukin 6: 6.4% ± 2.95% vs 1.7% ± 0.63%, p = 0.055). In addition, mesothelin-targeting chimeric antigen receptor–modified T cells showed greater inhibitory and proliferative capability than control T cells within EH-CA1a cell xenografts. This study shows the potential of mesothelin-targeting chimeric antigen receptor–modified T cells in treating bile duct carcinoma.
Introduction
Bile duct carcinoma (BDC), a devastating malignancy arising from the bile duct epithelial cells, is the second most common primary hepatobiliary malignant disease, and with 0.6 to 1 of 100,000 individuals in the United States per year.1,2 It is a rare malignancy of the biliary tract, and it is difficult to diagnose and associated with a high mortality. 3 Current traditional treatments such as surgical resection, chemotherapy, and radiotherapy are failing to reduce the recurrence rate or increase the 5-year survival rate of BDC patients.4,5 Thus, novel treating modalities for BDC patients are in desperate need.
Chimeric antigen receptor (CAR) modified T cell–based immunotherapy has achieved outstanding success in treating cancers, especially B-cell hematologic malignancies.6–8 The reported average complete remission rate for treating acute lymphoblastic leukemia (ALL) and overall response rate for treating chronic lymphocytic leukemia (CLL) patients with CD19-CAR-T cells are 90.75% 9 and 65.83%, 10 respectively. The major two attributions for the success of treating B-cell hematologic malignancies with CAR-T cells are (1) sufficient antigen engagement facilitated by the distribution of malignant cells and (2) tolerable B-cell depletion–related side effects caused by broad CD19 expression on B cells.11,12 But unlike hematologic malignancies, treating solid tumors with CAR-T cells faces many obstacles, including immunosuppressive microenvironment within solid tumors and scarcity of CD19-like antigens.13,14 In addition, the transfection rate during manufacturing genetic engineered T cells is another determinant for the therapeutic effect of CAR-T cell–based immunotherapy.6,15
Mesothelin is a transmembrane glycoprotein with initial weight ~70 kDa before cleavage, during which mesothelin is processed into two fragments: ~40 kDa membrane-anchored C-terminal and ~32 kDa soluble N-terminal.16,17 The aberrant expression of mesothelin has been broadly found in a bunch of solid tumors, and this has been recently summarized in a review. 18 It has been found that mesothelin is highly expressed in bile duct cancer tissues, positive in 37% (7 of 19) of BDC and 100% (12 of 12) in common bile duct adenocarcinoma detected by immunohistochemistry.19–21 The expression of mesothelin was related to an unfavorable patient outcome and shorter patient survival of patients with extrahepatic bile duct cancer, ovarian cancer, and pancreatic cancer.22–24 In addition to cancer cells, mesothelin is also expressed on mesothelial cells of the peritoneal cavity, pleural cavity, and pericardium, as well as some other tissues, including rete testis and ovaries at a relatively lower level. 18 Hence, when treating mesothelioma, pancreatic cancer, ovarian cancer, and other solid tumors with mesothelin-targeting chimeric antigen receptor-modified T (mesoCAR-T) cells, the on-target/off-tumor toxicity induced by the expression of mesothelin on normal tissues should be taken into consideration. Nevertheless, it is reasonable to speculate that the side effects of mesoCAR-T cells treatment are somehow tolerable to certain group of patients; that is, side effect on testis is acceptable to male patients and side effect on ovary is acceptable to female patients. Thus, regarding its expression profile, that is, low expression level on normal cells and high expression level on various cancer cells, mesothelin is considered as a promising target for CAR-T cell–based immunotherapies. 18
PiggyBac, derived from the Trichoplusia ni genome with a large cargo capacity, is an important tool for genome engineering. 25 Compared to viral and other transgenic tools, the piggyBac transposon system possesses many advantages: (1) no viral effect concern, (2) large cargo capacity, (3) high efficient and stable integration, (4) a propensity to integrate into transcriptional units with non-random profile, and (5) lower genotoxicity.26,27
In this study, we developed the second-generation mesoCAR-T cells based on the piggyBac transposon system and investigated their potential in treating BDC. MesoCAR-T cells efficiently killed mesothelin-expressing EH-CA1a, EH-CA1b, and 293-meso cells while sparing mesothelin-negative GSC-SD, EH-GB1, L02, and control 293 cells in vitro. The in vivo assays revealed that mesoCAR-T cells inhibited the growth of mesothelin-expressing EH-CA1a cell–formed xenografts in the mouse model. Our results further propose mesoCAR-T cells for the treatment of BDC.
Materials and methods
Cell lines
Human cell lines K562 and HEK293 were purchased from American Type Culture Collection (ATCC, USA), GSC-SD and normal liver cell line L02 were purchased from the Chinese Academy of Sciences (China), and EH-GB1, EH-CA1a, and EH-CA1b were established by our laboratory.28,29 The EH-GB1 cells were derived from primary tumor tissue of a metastatic gallbladder carcinoma patient, and EH-CA1a and EH-CA1b cells were derived from a same primary tumor tissue of a pathologically proven BDC. For specificity controls, K562 and HEK293 cells were electroporated to express human mesothelin (designated as K562-meso and 293-meso, respectively). These cells were maintained in RPMI-1640 or RPMI–DMEM (Dulbecco’s Modified Eagle Medium) supplemented with 10% fetal bovine serum (FBS; Gibco, USA). All cells were incubated under 37°C, 5% CO2 conditions.
Real-time polymerase chain reaction
Total cellular RNA was isolated using TRIzol reagent (Invitrogen, USA; Life Technologies, USA). Complementary DNA (cDNA) was synthesized using the Quantscript RT Kit (TIANGEN, China) with 1.0 µg RNA in a 20-µL reaction system. Real-time polymerase chain reaction (RT-PCR) assays were carried out with SYBR Green PCR Master Mix (Toyobo, Japan). The polymerase chain reaction (PCR) running program was scheduled as follows: 95°C for 10 min, followed by 40 cycles of 95°C for 5 s, 60°C for 15 s, and 72°C for 5 s. The relative messenger RNA (mRNA) expression level was normalized to an endogenous control β-actin. Genomic DNA of T cells was extracted with a cell genome extraction kit (TIANGEN, China). RT-PCR assays were carried out by using the TaqMan probes according to the instruction. All reactions were performed in triplicate. The sequences of the primers used in this study are listed in Table 1.
Primer sequences used in this study.
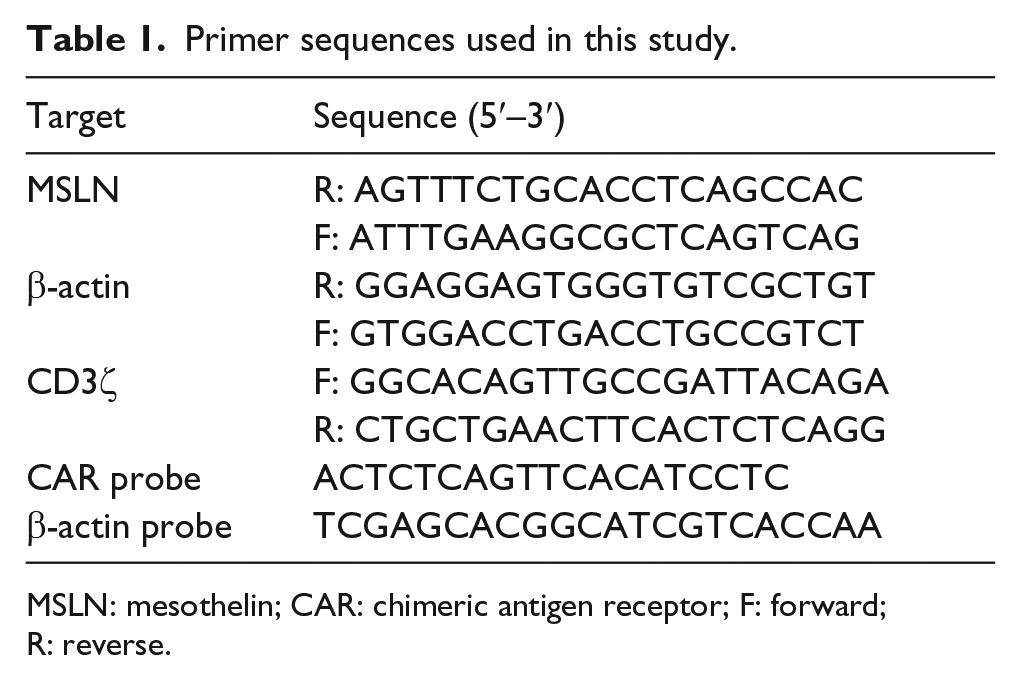
MSLN: mesothelin; CAR: chimeric antigen receptor; F: forward; R: reverse.
Western blot
The six-well plates were incubated on ice for 10 min with lysis buffer when cells covered approximately 90% of the bottom and then centrifuged at 12,000g, 4°C, to collect the total protein. Western blot assays were performed according to a previously described procedure. 30 The primary antibodies used were mouse anti-mesothelin (Cell Signaling Technology (CST), USA), mouse anti–glyceraldehyde 3-phosphate dehydrogenase (GAPDH; Beyotime, China), and mouse anti-CD3ζ (Abcam, UK). The secondary antibody was horseradish peroxidase (HRP)-labeled goat anti-mouse immunoglobulin G (IgG) (H+L; Beyotime). The expression of each band was quantitatively analyzed using the Image Lab™ software (Bio-Rad, USA) and normalized to the expression of GAPDH in the same lane.
MesoCAR-T cell manufacturing
Peripheral blood mononuclear cells (PBMCs) were obtained by Ficoll density gradient centrifugation and then placed in an incubator for 2–4 h. About 5 × 106 floating cells were collected after 2–4 h of incubation, washed with phosphate-buffered saline (PBS) twice, and then electric transferred with 6 µg mesoCAR plasmid or empty buffer by an electroporator (Lonza, Switzerland) with the Amaxa® Human T Cell Nucleofector® Kit. T cells were cultured in AIM-V medium (Gibco) containing anti-CD3 and anti-CD28 antibodies and stimulated by the addition of interleukin (IL)-2 (500 IU/mL) and IL-15 (10 ng/mL).
Enzyme-linked immunosorbent assay
Cytokine release assays were performed after co-culturing 5 × 105 T cells with 5 × 105 target cells within 3 mL medium per well in six-well plates. After 24 h, cell-free supernatants were collected to detect the concentration of IL-2, IL-4, IL-6, IL-10, tumor necrosis factor α (TNF-α), and interferon γ (IFN-γ) with BD™ Cytometric Bead Array (CBA) Human Th1/Th2 Cytokine Kit II according to the manufacturer’s instruction. The concentration of IFN-γ was also detected by an enzyme-linked immunosorbent assay (ELISA) kit (Dakewe, China). Values represent the mean of triplicate wells.
Flow cytometry
MesoCAR expression in mesoCAR-T cells was detected by using fluorescein isothiocyanate (FITC)-conjugated anti-mouse F(ab′)2 goat IgG (CST). The expression of mesothelin on the surface of aforementioned tumor cells was detected by using mouse anti-human mesothelin antibody. T-cell phenotypes were determined with monoclonal antibodies for CD28 (Beckman, USA), CD137, CD134, CD27, and CD107α (Biosciences (BD), USA). Intracellular cytokines were determined with monoclonal antibodies for IFN-γ, TNF-α, IL-2, and IL-10, following incubation with cytokines release stimulator for 4–6 h, and processed with the IntraPrep Permeabilization Reagent (Beckman). Flow cytometric analysis was performed on a BD FACS Caliber, and the data were analyzed with FlowJo software.
Cytotoxicity assays
The cytotoxicity of mesoCAR-T cells was measured in real time using an xCELLigence RTCA SP label-free, impedance-based cell sensing device (Roche Applied Science, Canada). The L02, EH-CA1b, GSC-SD, and EH-GB1 cells (1 × 104 cells per well) were suspended in DMEM or RPMI-1640 media containing 10% FBS (Gibco). The cell suspensions were transferred to 16-well microtiter plates incorporating a sensor electrode array (E-plates). Cell proliferation was measured as a change in relative impedance, termed cell index (CI). After 24 h, the effector cells were added at the ratio of effector to target cell (E:T = 8:1, 4:1, and 2:1) in a total volume of 150 µL. The cytotoxicity was then monitored by measuring changes in impedance as CI values recorded by the xCELLigence RTCA SP device. The cytotoxicity of mesoCAR-T cells was also determined by Cytotoxicity LDH (lactic dehydrogenase) Assay Kit-WST (Dojindo, Japan) by co-culturing with floating EH-CA1a and K562-meso cells, in which 4 × 103 target cells were co-cultured with mesoCAR-T cells (E:T = 8:1, 4:1, and 2:1) for 6 h.
In vivo tumorigenesis assay
All mice used in this study were approved by the Medical Experimental Animal Care Unit of the Second Military Medical University. Nonobese diabetic/severe combined immunodeficiency (NOD/SCID) mice aged 5–6 weeks were purchased from the Shanghai SLAC Laboratory Animal Co. Ltd of Chinese Academy of Sciences, and the mice were maintained in a specific-pathogen-free (SPF) condition. A total of 1 × 107 EH-CA1a-Luc cells were subcutaneously transplanted into mice. After 10 days, tumor implantation was confirmed by bioluminescence images (BLIs) using a Xenogen IVIS imaging system (Life Technologies), and six tumor-bearing mice per group were randomized before treatment. Then, 5 × 106 mesoCAR-T cells, control T cells, and PBS were injected intratumorally at day 0. After 7 days, 5 × 106 mesoCAR-T, control T cells, and PBS were injected again. The tumor BLI was quantified once a week after CAR-T cells injection for 30 days.
Statistical analysis
All data are presented as the mean ± standard deviation. Independent Student’s t-test was used to analyze the variation of two selected groups, and Pearson’s chi-square test was used to analyze the correlation of two parameters. Values of p <0.05 were considered statistically significant, and values of p <0.01 were considered highly statistically significant. All statistical analyses were performed with SPSS (Statistical Package for the Social Sciences) version 18.0 software.
Results
Development of mesoCAR-T cells by piggyBac transposon system
With regard to the therapeutic improvement of the second-generation CAR-T cells compared to the first-generation CAR-T cells owing to the incorporation of co-stimulatory domains, 31 we developed the second-generation mesoCAR-T cells with a co-stimulatory module from 4-1BB molecule by the piggyBac transposon system (Figure 1(a)). Primary human T cells were electroporated with mesoCARs, and the control T cells were electroporated without mesoCARs. Western blot assay showed that both mesoCAR-T cells and control T cells expressed ~17 kDa endogenous CD3ζ, while the expression of the ~69 kDa exogenous CD3ζ was peculiar to mesoCAR-T cells (Figure 1(b)). RT-PCR assay with the usage of specific primers targeting exogenous CD3ζ further confirmed that the expression of the exogenous CD3ζ was peculiar to mesoCAR-T cells (Figure 1(c)). In addition, flow cytometry (FCM) assay showed that the expression of mesoCARs was not detectable in control T cells, while efficiently expressed by 66.6% of mesoCAR-T cells (Figure 1(d)). These results indicated the success in the development of mesoCAR-T cells.

Development of mesoCAR-T cells. (a) Linearized depiction of mesoCAR vector with 4-1BB co-stimulatory molecule module. (b) The expression of exogenous CD3ζ was tested by western blot assay; the 69 kDa weight band of exogenous CD3ζ was detected in mesoCAR-T cell lane instead of control T cell lane; M, marker. (c) The expression of exogenous CD3ζ was only found in mesoCAR-T cells by RT-PCR assay with specific primers targeting exogenous CD3ζ. (d) FCM assay with FITC-labeled goat anti-mouse IgG F(ab′)2 antibody revealed mesoCAR expression from 66.0% of mesoCAR-T cells, while ignorable in control T cells.
Cell phenotype of mesoCAR-T cells
Thereafter, the confirmation on the expression of mesoCARs by transduced mesoCAR-T cells motivated us to investigate the functional state of mesoCAR-T cells. By co-culture with mesothelin-expressing K562 (K562-meso) cells, the morphology of activated T cells has been indicated different with inactivated T cells, with the change from a formal round shape into a bigger shuttle-like shape (Figure 2(a)). Then, the surface markers of CD107α, CD27, CD28, CD137, and CD134 were detected by flow cytometry assays. All CD107α, CD27, CD137, CD28, and CD134 were expressed by mesoCAR-T cells at higher level compared to control T cells (CD107α: 71.9% vs 48.6%; CD27: 92.1% vs 61.8%; CD137: 55.5% vs 8.4%; CD28: 98.0% vs 82.1%; CD134: 37.5% vs 10.4%; Figure 2(b)). These results indicated that control T cells were insensitive to K562-meso cells, while mesoCAR-T cells were effectively activated by K562-meso cells.

The cell phenotype of mesoCAR-T cells. (a) mesoCAR-T cells changed from a formal round shape into a shuttle-like shape after co-culture with K562-meso cells. (b) Activation markers CD107α, CD27, CD137, CD28, and CD134 were tested by FCM; all these markers were expressed by mesoCAR-T cells at higher level than control T cells (CD107α: 71.9% vs 48.6%; CD27: 92.1% vs 61.8%; CD137: 55.5% vs 8.4%; CD28: 98.0% vs 82.1%; CD134: 37.5% vs 10.4%).
MesoCAR-T cells exert cytotoxic effect toward mesothelin-expressing BDC cells
We next investigated the cytotoxicity of mesoCAR-T cells toward target cells. A line of tumor cells were used to detect mesothelin expression by FCM assay, and results showed that BDC EH-CA1b and EH-CA1a cells were positive for mesothelin expression (30.7% and 78.9%, respectively), while GSC-SD, EH-GB1, and normal liver L02 cells were relatively negative for mesothelin expression (7.8%, 8.9%, and 4.9%, respectively) (Figure 3(a)). The mesothelin expression at protein level of EH-CA1a, EH-CA1b, GSC-SD, and EH-GB1 cells was further tested by western blot assay (Figure 3(b)). Thus, EH-CA1a and EH-CA1b cells were chosen as mesothelin-positive target tumor cells to test the function of mesoCAR-T cells, while GSC-SD, EH-GB1, and L02 cells were chosen as mesothelin-negative control tumor cells. The xCELLigence RTCA DP Instrument was applied to test the capability of mesoCAR-T cells to kill target tumor cells by co-culturing at different E:T ratios (8:1, 4:1, and 2:1 for mesoCAR-T cells and 8:1 for control T cells). Data showed that mesoCAR-T cells exhibited significant cytotoxic activity toward mesothelin-positive EH-CA1b cells in an E:T ratio-dependent manner, while sparing mesothelin-negative GSC-SD, EH-GB1, and L02 cells, whereas control T cells exerted almost no killing effect toward all four cell lines at E:T = 8:1 (Figure 3(c)–(d)). The cytotoxicity of mesoCAR-T cells toward mesothelin-positive tumor cell EH-CA1a was confirmed by LDH assay; results showed that the lysis rate of tumor cells achieved 40% at E:T = 2:1, while control T cells exerted almost no cytotoxicity toward EH-CA1a cells even at E:T = 8:1 (Figure 3(e)). In addition, ELISA was applied to test the cytokine secretion of mesoCAR-T cells post co-culture with these four tumor cell lines. Results showed that mesothelin-positive EH-CA1b and EH-CA1a cells induced much higher level of IL-2, IL-6, TNF-α, and IFN-γ secretion than mesothelin-negative GSC-SD and EH-GB1 cells from mesoCAR-T cells (IL-2: 823.4 ± 105.4 and 1230.3 ± 138.9 vs 251.9 ± 124.9 and 237.3 ± 59.9 pg/mL; IL-6: 381.5 ± 112.5 and 582.7 ± 68.8 vs 180.7 ± 47.6 and 186.7 ± 57.3 pg/mL; TNF-α: 451.8 ± 139.1 and 668.9 ± 88.2 vs 144.7 ± 79.4 and 131.6 ± 67.7 pg/mL; IFN-γ: 15,815.6 ± 684.7 and 30,956.7 ± 846.9 vs 7933.9 ± 382.8 and 8120.7 ± 282.6 pg/mL), while a comparable amount of IL-4 and IL-10 was found (IL-4: 254.8 ± 42.9 and 283.6 ± 53.4 vs 241.6 ± 48.5 and 218.4 ± 31.4 pg/mL; IL-10: 551.4 ± 82.2 and 626.0 ± 67.8 vs 546.6 ± 32.5 and 486.1 ± 28.2 pg/mL) (Figure 3(f)). These results indicated that mesoCAR-T cells specifically kill mesothelin-expressing BDC cells while sparing mesothelin-negative gallbladder cancer cells.

MesoCAR-T cells were cytotoxic toward mesothelin-expressing BDC cells. (a) A line of tumor cell lines were examined for the expression of mesothelin by FCM assay, and showed variable level among tumor cell lines. (b) Western blot assay revealed 40 kDa mesothelin-positive EH-CA1b and EH-CA1a and mesothelin-negative GSC-SD and EH-GB1 cells. Mesothelin precursor band of ~70 kDa was also detected; GAPDH was used as endogenous control. (c) xCELLigence RTCA DP cytotoxic equipment data showed that mesoCAR-T cells killed EH-CA1b cells in an E:T ratio (8:1, 4:1, and 2:1)–dependent manner, while sparing L02, GSC-SD, and EH-GB1 cells, whereas control T cells barely showed cytotoxicity toward all four cell lines at E:T = 8:1. (d) The xCELLigence RTCA DP cytotoxic equipment data were extracted and calculated into specific lytic percentage of EH-CA1b, L02, GSC-SD, and EH-GB1 cells by mesoCAR-T cells. (e) Lactic dehydrogenase (LDH) release assay was applied to further confirm the cytotoxicity of mesoCAR-T and control T cells against EH-CA1a. Results showed that mesoCAR-T cells lysed mesothelin-positive EH-CA1a cells, while control T cells exerted almost no cytotoxicity toward EH-CA1a cells at E:T = 8:1. (f) EH-CA1a and EH-CA1b cells were more potent in inducing IL-2, IL-6, TNF-α, and IFN-γ secretion than GSC-SD and EH-GB1 cells of mesoCAR-T cells.
MesoCAR-T cells eliminate 293-meso and K562-meso cells
In order to confirm the cytotoxicity of mesoCAR-T cells toward target cells and exclude tumor cell–related killing effect, mesothelin-negative HEK293 cells were genetically manipulated to express mesothelin. RT-PCR assay was first applied to detect mesothelin expression after electroporation of 293 cells with mesothelin-expressing vector, and showed an efficient expression of mesothelin from 293-meso cells (Figure 4(a)). The xCELLigence RTCA DP cytotoxic equipment data showed that mesoCAR-T cells were cytotoxic toward 293-meso cells in an E:T ratio-dependent manner, while control T cells did not inhibit the growth of 293-meso cells (Figure 4(b)). The secretion of IFN-γ from mesoCAR-T cells post co-culture with 293 and 293-meso cells was also tested by ELISA and showed improvement on the ability of 293-meso cells in inducing mesoCAR-T cells to secrete IFN-γ compared to 293 cells (1458.8 ± 69.01 vs 81.32 ± 3.33 pg/mL). Then, the intracellular expressions of IFN-γ, TNF-α, IL-2, and IL-6 were also detected by FCM assay after co-culture of 293-meso cells with control T and mesoCAR-T cells, and results showed significant improvement on the expression of these cytokines of mesoCAR-T cells compared to control T cells (IFN-γ: 85.1% ± 1.47% vs 8.3% ± 2.50%, p = 0.000; TNF-α: 90.9% ± 4.67% vs 18.5% ± 3.62%, p = 0.0004; IL-2: 60.8% ± 2.00% vs 15.6% ± 2.06%, p = 0.002; IL-6: 6.4% ± 2.95% vs 1.7% ± 0.63%, p = 0.055; Figure 4(c)).
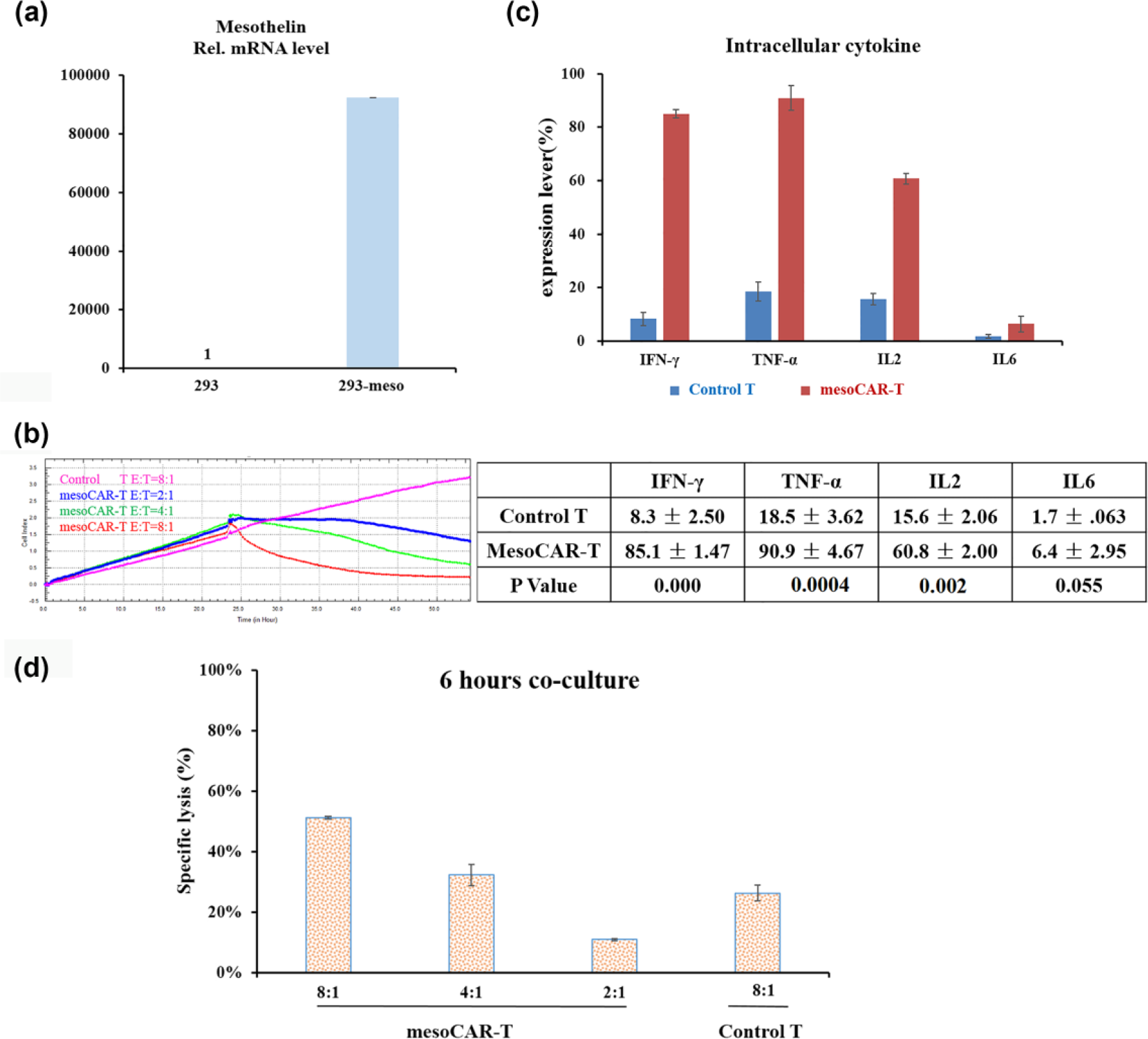
MesoCAR-T cells eliminate 293-meso cells. (a) Mesothelin expression of 293-meso cells was detected by RT-PCR with specific primers to transmembrane mesothelin fragment after electroporation of 293 cells with mesothelin-expressing plasmid. (b) The xCELLigence RTCA DP cytotoxic equipment data revealed an E:T ratio-dependent cytotoxic effect of mesoCAR-T cells toward 293-meso cells, while control T cells did not inhibit the growth of 293-meso cells at E:T = 8:1. (c) FCM assay showed that 293-meso cells were able to induce a higher expression level of IFN-γ, TNF-α, IL-2, and IL-6 than control T cells from mesoCAR-T cells (IFN-γ: 85.1% ± 1.47% vs 8.3% ± 2.50%, p = 0.000; TNF-α: 90.9% ± 4.67% vs 18.5% ± 3.62%, p = 0.0004; IL-2: 60.8% ± 2.00% vs 15.6% ± 2.06%, p = 0.002; IL-6: 6.4% ± 2.95% vs 1.7% ± 0.63%, p = 0.055). (d) MesoCAR-T cells lysed 51%, 32%, and 11% of K562-meso cells at E:T = 8:1, 4:1, and 2:1, respectively, and control T cells lysed 26% of K562-meso cells at E:T = 8:1.
Moreover, the floating K562-meso feeder cells were also used to test the killing effect of control T cells and mesoCAR-T cells with LDH assay by co-culturing for 6 h, and results showed that mesoCAR-T cells lysed 51%, 32%, and 11% of K562-meso cells at E:T = 8:1, 4:1, and 2:1, respectively, while control T cells lysed only 26% of K562-meso cells at E:T = 8:1 (Figure 4(d)). These results further confirmed the cytotoxicity of mesoCAR-T cells toward mesothelin-expressing cells.
MesoCAR-T cells inhibit BDC xenograft growth in mouse model
MesoCAR-T cells inhibited the growth of EH-CA1a and EH-CA1b cells in vitro. We then investigated the effect of mesoCAR-T cells on the BDC cells in vivo. EH-CA1a-Luc cells were subcutaneously transplanted into NOD/SCID mice. Intratumoral injection of mesoCAR-T cells, control T cells, and PBS was conducted twice in 10 and 17 days post transplantation. Tumor burden was measured by BLIs with the Xenogen IVIS imaging system at days 0, 7, 14, 21, and 28 since the treatment. Results showed that control T cells barely inhibited the growth of xenografts, which is similar with PBS treatment, while mesoCAR-T cells exerted potent inhibition on the growth of xenografts (Figure 5(a) and (b)). All mice were sacrificed at day 28; xenograft tissues were exposed and weighted, and results showed that xenograft tissues from the mesoCAR-T cell–treated group were significantly smaller than the other two groups (Figure 5(c)). The proliferation of infused T cells was examined by RT-PCR with specific primers targeting endogenous CD3ζ gene, and results indicated that the endogenous CD3ζ gene amount of mesoCAR-T cell–treated xenografts was about 40 times of that of control T cell–treated xenografts (Figure 5(d)), which indicated that mesoCAR-T cells proliferated greater than control T cells in xenograft tissues. In summary, these results indicated that mesoCAR-T cells were able to inhibit the growth of mesothelin-positive xenografts in vivo.

MesoCAR-T cells inhibit the growth of mesothelin-positive EH-CA1a xenografts in vivo. (a) EH-CA1a-Luc cells were subcutaneously injected into NOD/SCID mice at 1 × 107 cells per mouse. Ten days later, the mice were treated with control T cells or mesoCAR-T cells at 5 × 106 cells per mouse (day 0); tumor growth was quantified by BLI imaging once a week after the first CAR-T and control T cells injection. A repeated treatment was carried out at day 7 with 5 × 106 cells per mouse of the same cells. (b) Tumor growth curve: p = 0.0386 when the mesoCAR-T treatment group was compared to the PBS group, and p = 0.0223 when the mesoCAR-T treatment group was compared to the control T treatment group. (c) After 28 days, all mice were sacrificed by diethyl ether. The tumor weight of each group was analyzed by Graphpad Prism 6.0, p = 0.027 when mesoCAR-T treatment group was compared to PBS treatment group. Values are represented as mean ± SD. (d) RT-PCR with primers specifically targeting endogenous CD3ζ gene revealed that mesoCAR-T cells proliferated as ~40 times as control T cells within xenograft tissues.
Discussion
In this study, we explored mesoCAR-T cell–based immunotherapy to treat BDC based on the piggyBac transposon system. The results indicated that our modified piggyBac mediated approximately 66.0% T cells expressing mesoCAR, which could exhibit comparable efficiency in transducing CAR expression with lentiviral and retroviral systems. The results from the in vitro and in vivo functional investigations of mesoCAR-T cells toward EH-CA1a and EH-CA1b BDC cells provided evidence for further pre-clinical and clinical studies of mesoCAR-T cells for the treatment of BDC. The potential of mesoCAR-T cells has been investigated in treating pancreatic cancer (NCT01897415, NCT02465983), mesothelioma (NCT01355965, NCT02414269), and metastatic lung and breast cancers (NCT02414269). While to our knowledge, this is the first report investigating the potential of mesoCAR-T cells in the treatment of BDC.
Mesothelin is expressed on normal mesothelial cells in the pleura, pericardium, and peritoneum, raising the concerns about on-target/off-tumor toxicity of mesoCAR-T cells.32,33 Previously, the use of high doses of ErbB2 CAR-T cells has resulted in a fatal adverse event, attributed by the low-level ErbB2 expression in healthy lung epithelial and cardiovascular cells. 34 The risk of on-target/off-tumor toxicity has led to different strategies: (1) fine-tuning the affinity of single-chain variable fragment (scFv) antibody of CARs, to reduce the sensibility of CAR-T cells and lead them to specifically kill tumor cells with higher antigen expression 35 ; (2) introduction of suicide genes; for example, inducible caspase-9 system is interposed into CAR gene, and CAR-T cells can be depleted by the administration of a synthetic small-molecule drug 36 ; (3) dual receptors system; the binding of a receptor to A antigen drives the inducible expression of the other CAR for B antigen; hence, the modified T cells are only activated in the presence of both antigens 37 ; and (4) transiently expressed CARs; the function of CAR-T cells is transiently obtained by transfection of CAR mRNA. 38 Thus, a suitable strategy should be adopted before the mesoCAR-T cell–based immunotherapy enters into the clinical trial.
In the in vitro studies, control T cells were found partially activated by anti-CD3 and anti-CD28 antibodies containing cytokine cocktail. This may be resulted from the non-specific activating effect of anti-CD3 and anti-CD28 antibodies (cytokine-induced killer cell manufacturing procedure), 39 which is in consistency with the comparable CD28 expression of control T cells with mesoCAR-T cells (82.1% vs 98.0%) and the distinct cytotoxicity toward EH-CA1a and EH-CA1b cells between control T cells and mesoCAR-T cells.
Potent proliferation capability within EH-CA1a xenografts in the mouse model was observed in mesoCAR-T cells instead of control T cells. There are two most possible attributes to this difference: (1) Control T cells could not be activated by mesothelin-expressing EH-CA1a cells in vivo, though partially non-specific activated cytokine cocktail in vitro; mesoCAR-T cells could be activated by EH-CA1a cells through mesothelin engagement and (2) the incorporation of the 4-1BB co-stimulatory module might reprogram our second-generation mesoCAR-T cells into a central memory differentiation state with enhanced respiratory capacity and persistence in vivo. 40
However, our mesoCAR-T cells failed to eliminate targeting cells entirely in vitro or in vivo. There are many possible reasons for this failure: (1) the expression distribution of mesothelin in BDC cells, (2) the soluble mesothelin fragments after cleavage may stand for another factor impacting the therapeutic effect of mesoCAR-T cells immunotherapy, 18 and (3) the microenvironment within xenografts suppressed mesoCAR-T cells.
In summary, we developed mesoCAR-T cells through our modified piggyBac transposon system with high transducing efficiency. Our mesoCAR-T cells exhibited potent cytotoxicity toward mesothelin-expressing EH-CA1a and EH-CA1b cells in vitro and in vivo. Thus, this study holds great potential for the treatment of mesothelin-positive solid tumors.
Footnotes
Acknowledgements
J.Y.X, Z.L.Y, and D.Q.J contributed equally to this article.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
This work was supported by the National Science Fund Projects (81672997, 31601075) and the Capacity Building Project of Shanghai Engineering Research Center (16DZ2281000).